Xenium Dimensionality Reduction with or without normalization and transformation

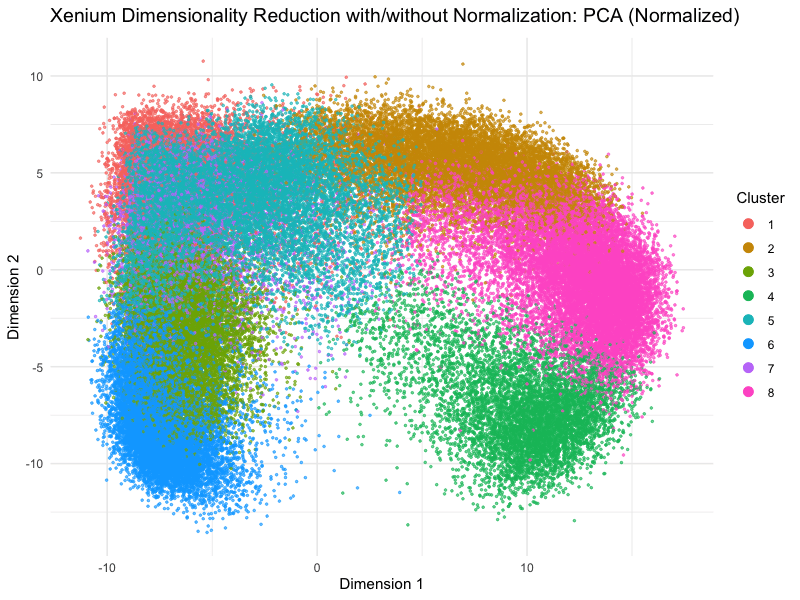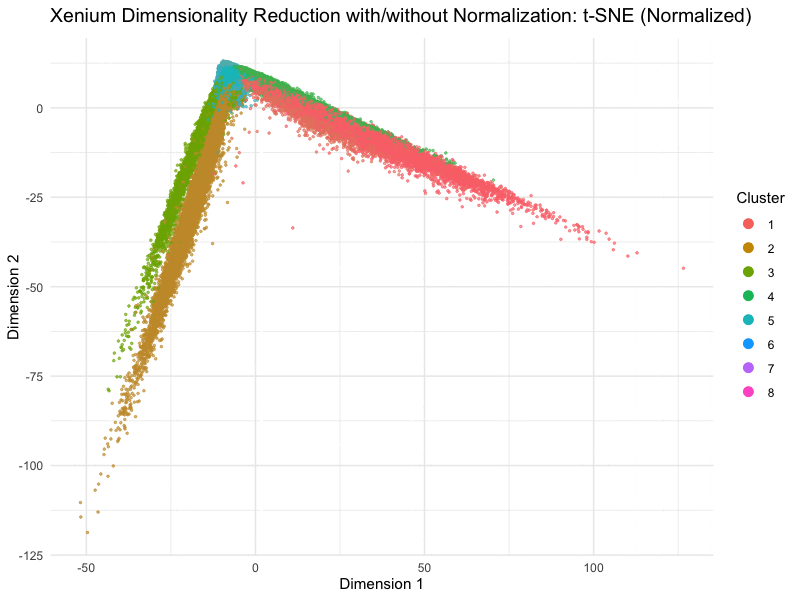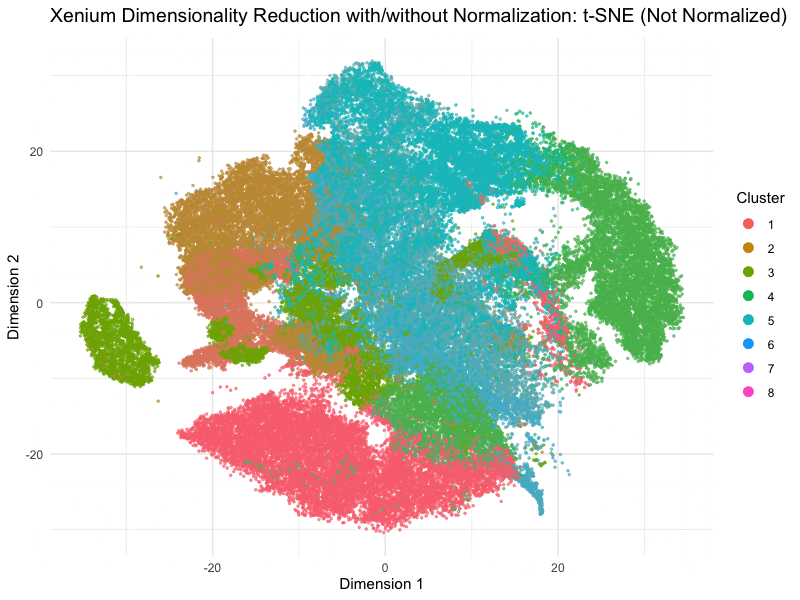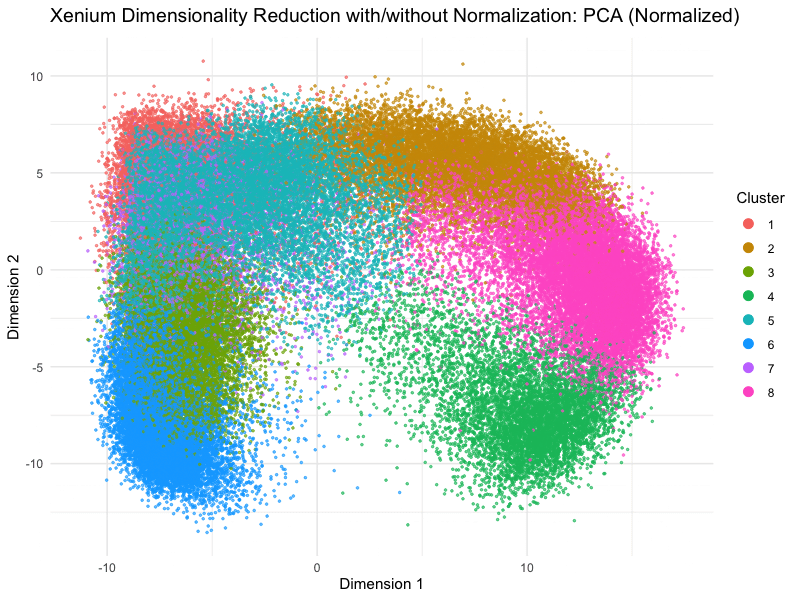
This animation depicts how normalization and log-transformation steps are essential in accurate dimensionality reduction that allows biological interpretation rather than being obscured in noise. Without normalization, the PCA space appears as two lines orthogonal to each other in the 2D space. This likely means that our PC1 and PC2 was heavily impacted by the total counts and sizes of the cell due to total expression. Similarly, the tSNE without normalization does not depict distinct islands because the distances between neighborhoods are skewed by outliers, a disadvantage of no longer having a normalized, gaussian distribution. On the other hand, with log-transformation and normalization, we are able to truly see the variance between gene expression. The data in the PCA space now has much more depth than the initial V shape, allowing us to distinguish between clusters and their variance.
On a more technical aspect, the legend remains visible at the right side. View follow allows the scale of axes to change between plots. Axes titles are generalized to dimension 1 and 2 as compared to tSNE1 or PC1. Plot titles change between graphs as well for easier interpretation. Beyond the visualization, scree plots and elbow plots were made to help decide PC numbers and clusters between the data. Data without normalization warranted lower number of clusters, likely due to true neighborhoods being obscured by the noise generated by PC loadings being affected by cell size and count. This shows how normalization allowed higher resolution of our data analysis, because previously, the cells may have been grouped into undefined populations because of outliers such as many lowly expressed genes or a few extremely highly expressed outliers.
Code
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
library(ggplot2)
library(gganimate)
library(Rtsne)
library(patchwork)
data <- read.csv('~/Desktop/GDV/Xenium-IRI-ShamR_matrix.csv.gz')
pos <- data[,c('x','y')]
rownames(pos)<-data[,1]
gexp<-data[,4:ncol(data)]
rownames(gexp)<-data[,1]
head(pos)
gexp[1:5,1:5]
rownames(gexp) <- data[,1]
totgexp <- rowSums(gexp)
#normalizing the data
mat <- log10(gexp/totgexp * 1e6 + 1)
dim(mat)
#PCA
pcs_raw <- prcomp(gexp, center = TRUE, scale = FALSE)
plot(pcs_raw$sdev[1:50]) #first 7 pcs chosen based on this plot
# Determining optimal k
wss <- sapply(1:15, function(k){kmeans(pcs_raw$x[,1:7], centers=k, nstart=10)$tot.withinss})
elbow_df <- data.frame(k=1:15, wss=wss)
p_elbow <- ggplot(elbow_df, aes(x=k, y=wss)) +
geom_line() +
geom_point() +
theme_minimal() +
labs(title="Determining Optimal clusters", x="Number of Clusters (k)", y="Total within sum of squares") +
theme(plot.title = element_text(size = 9, face = "bold"))
p_elbow
clusters_raw <- as.factor(kmeans(pcs$x[,1:7], centers = 5)$cluster)
df_clusters_raw <- data.frame(pos, emb_raw, pcs_raw$x[,1:7], cluster = clusters_raw)
pcs_norm <- prcomp(mat, center = TRUE, scale = FALSE)
plot(pcs_norm$sdev[1:50]) #first 10 pcs chosen based on this plot
# Determining optimal k
wss <- sapply(1:15, function(k){kmeans(pcs_norm$x[,1:10], centers=k, nstart=10)$tot.withinss})
elbow_df <- data.frame(k=1:15, wss=wss)
p_elbow <- ggplot(elbow_df, aes(x=k, y=wss)) +
geom_line() +
geom_point() +
theme_minimal() +
labs(title="Determining Optimal clusters", x="Number of Clusters (k)", y="Total within sum of squares") +
theme(plot.title = element_text(size = 9, face = "bold"))
p_elbow
clusters_norm <- as.factor(kmeans(pcs_norm$x[,1:7], centers = 8)$cluster)
df_clusters_norm <- data.frame(pos, emb_norm, pcs_norm$x[,1:10], cluster = clusters_norm)
set.seed(42)
tsne_raw <- Rtsne::Rtsne(pcs_raw$x[, 1:7], dims=2, perplexity=30, verbose=TRUE) #verbose parameter added for troubleshooting
tsne_norm <- Rtsne::Rtsne(pcs_norm$x[, 1:10], dims=2, perplexity=30, verbose=TRUE)
emb_raw <- tsne_raw$Y
emb_norm <- tsne_norm$Y
rownames(emb_raw) <- rownames(mat)
colnames(emb_raw) <- c('tSNE1', 'tSNE2')
head(emb_raw)
rownames(emb_norm) <- rownames(mat)
colnames(emb_norm) <- c('tSNE1', 'tSNE2')
head(emb_norm)
#cluster in pcs
p1 <- ggplot(df_clusters_raw, aes(x=PC1, y=PC2, col=cluster)) +
geom_point(size = 0.5) +
theme_minimal() +
coord_fixed()+
labs(title="Clusters in PCA Space (Not normalized)") +
guides(color = guide_legend(override.aes = list(size = 2, alpha = 1))) +
theme(plot.title = element_text(size = 9, face = "bold"))
p1
#clusters in tsne
p2 <- ggplot(df_clusters_norm, aes(x=PC1, y=PC2, col=cluster)) +
geom_point(size = 0.5) +
theme_minimal() +
coord_fixed()+
labs(title="Clusters in PCA Space (Normalized)") +
guides(color = guide_legend(override.aes = list(size = 2, alpha = 1))) +
theme(plot.title = element_text(size = 9, face = "bold"))
p2
p3 <- ggplot(df_clusters_raw, aes(x=tSNE1, y=tSNE2, col=cluster)) +
geom_point(size = 0.5) +
theme_minimal() +
coord_fixed()+
labs(title="Clusters in tSNE Space (Not normalized)") +
guides(color = guide_legend(override.aes = list(size = 2, alpha = 1))) +
theme(plot.title = element_text(size = 9, face = "bold"))
p3
p4 <- ggplot(df_clusters_norm, aes(x=tSNE1, y=tSNE2, col=cluster)) +
geom_point(size = 0.5) +
theme_minimal() +
coord_fixed()+
labs(title="Clusters in tSNE Space (Normalized)") +
guides(color = guide_legend(override.aes = list(size = 2, alpha = 1))) +
theme(plot.title = element_text(size = 9, face = "bold"))
p4
(p1+p2)/(p3+p4)
#AI prompt: how do I gganimate between 4 plots that I have created
d1 <- data.frame(X = pcs_raw$x[,1], Y = pcs_raw$x[,2],
Stage = "PCA (Not Normalized)", Cluster = clusters_raw, ID = 1:nrow(mat))
d2 <- data.frame(X = pcs_norm$x[,1], Y = pcs_norm$x[,2],
Stage = "PCA (Normalized)", Cluster = clusters_norm, ID = 1:nrow(mat))
d3 <- data.frame(X = emb_raw[,1], Y = emb_raw[,2],
Stage = "t-SNE (Not Normalized)", Cluster = clusters_raw, ID = 1:nrow(mat))
d4 <- data.frame(X = emb_norm[,1], Y = emb_norm[,2],
Stage = "t-SNE (Normalized)", Cluster = clusters_norm, ID = 1:nrow(mat))
df_anim <- rbind(d1, d2, d3, d4)
anim <- ggplot(df_anim, aes(x = X, y = Y, color = Cluster, group = ID)) +
geom_point(size = 0.5, alpha = 0.6) +
theme_minimal() +
transition_states(Stage, transition_length = 3, state_length = 2) +
view_follow() +
labs(title = "Xenium Dimensionality Reduction with/without Normalization: {next_state}",
x = "Dimension 1", y = "Dimension 2") +
theme(plot.title = element_text(face = "bold", size = 14)) +
guides(color = guide_legend(override.aes = list(size = 3, alpha = 1))) +
ease_aes('cubic-in-out')
animate(anim, nframes = 200, fps = 20, width = 800, height = 600, res = 100)
anim_save("maanya_bajaj.gif")
#References - gganimate documentation