Deconvolution on Visium Dataset

Description
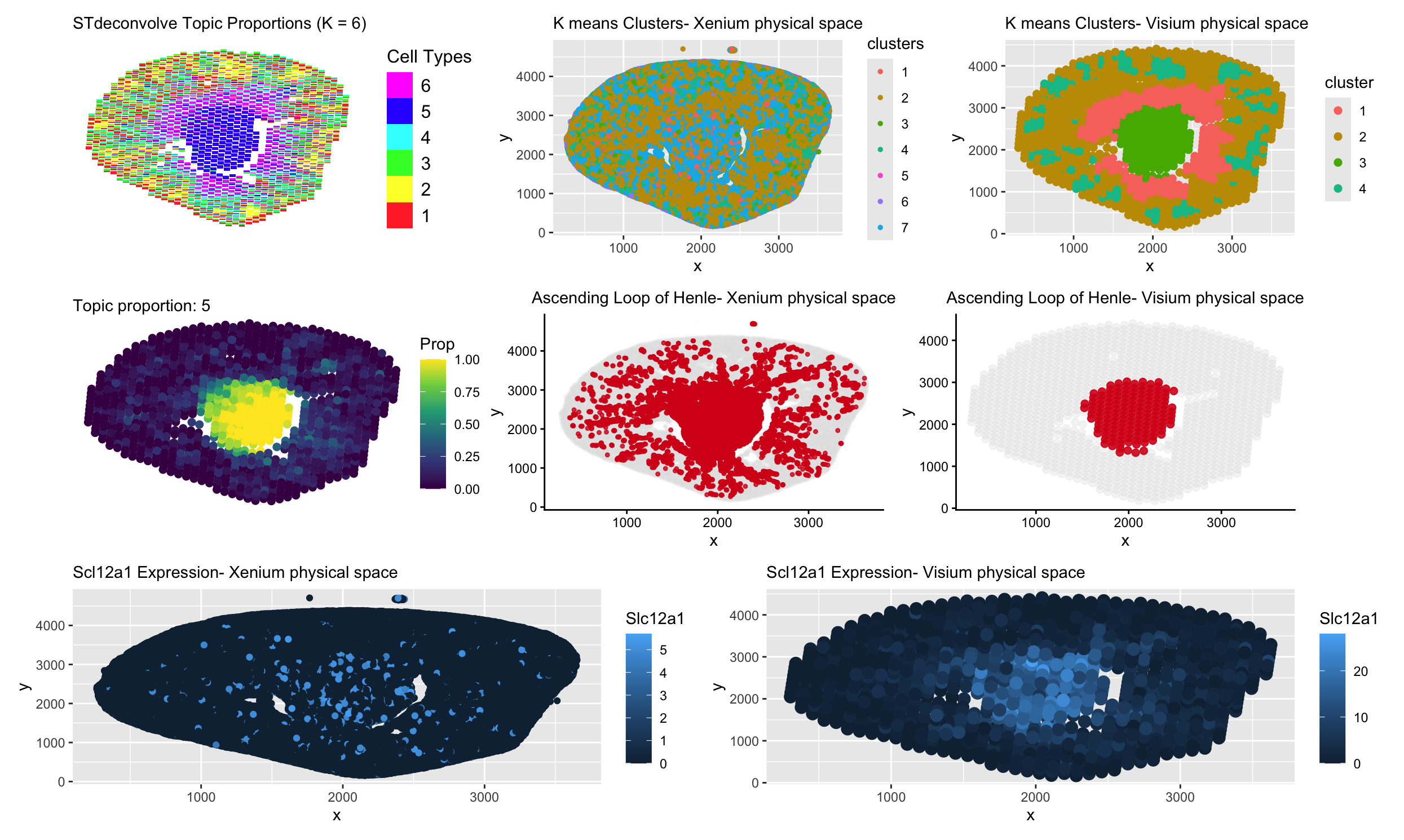
In the deconvolution analysis (Visium), the topic of interest looks very tightly concentrated in the center. Each Visium spot contains multiple cells. STdeconvolve models mixtures of transcriptional programs within each spot, so a topic only shows strong enrichment where that cell type makes up a large proportion of the spot. In the kidney, the Ascending Loop of Henle is organized in a fairly compact central region, so the deconvolved topic lights up clearly there. Outside that region, even if a few relevant cells are present, their signal gets diluted by other cell types within the same spot. As a result, the estimated proportion stays low and the enrichment looks very clean and centered.
In contrast, Xenium works at single-cell resolution. Each point is an individual cell, not a mixture. Cells from the Ascending Loop are arranged along tubular structures and can extend beyond one tightly packed area. Because clustering assigns each cell to a group independently, the cluster of interest looks more scattered and less strictly confined. What we see there is the true spatial spread of individual cells rather than an averaged signal across spots.
The same idea applies to the marker gene, Slc12a1. In Xenium, expression looks more distributed because you can detect every expressing cell wherever it sits. In Visium, expression is averaged across all cells in a spot, so strong signal only appears where many Slc12a1 positive cells are concentrated together. That makes the Visium signal look smoother and more centralized. Overall, the difference comes down to resolution, deconvolution and Visium gene expression reflect spot-level mixtures, while Xenium captures the spatial distribution of individual cells.
5. Code (paste your code in between the ``` symbols)
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
269
270
271
272
273
274
275
276
277
278
279
280
281
282
283
284
285
286
287
288
289
290
291
292
293
294
295
296
297
298
299
300
301
302
303
304
305
306
307
308
309
310
311
312
313
314
315
316
317
318
319
320
321
322
323
324
325
326
327
328
329
330
331
332
333
334
335
336
337
338
339
340
341
342
343
344
345
346
347
348
349
350
351
library(ggplot2)
library(patchwork)
require(remotes)
remotes::install_github('JEFworks-Lab/STdeconvolve')
library(STdeconvolve)
remotes::install_github('JEFworks-Lab/scatterbar')
library(scatterbar)
# # PLAN
# 1. Load xenium and perform clustering and diff expression analysis
# 2. Load visum and perform deconvolution and diff expression analysis
# 3. Get the interested cell type proportion of the two
# load Xenium data
data <- read.csv("~/Documents/genomic-data-visualization-2026/data/Xenium-IRI-ShamR_matrix.csv.gz")
# get the spatial positions
pos <- data[,c('x', 'y')]
rownames(pos) <- data[,1]
# get gene expression values
gexp <- data[, 4:ncol(data)]
rownames(gexp) <- data[,1]
# normalize
totgexp= rowSums(gexp)
mat <- log10(gexp/totgexp *1e6 +1)
# 2. linear dimensionality reduction
pcs <- prcomp(mat)
# visualize PC
df <- data.frame(
pos,
pcs$x
)
# # 3. tSNE
# ts <- Rtsne::Rtsne(pcs$x[,1:10], dim=2)
# names(ts)
# emb <- ts$Y
# df <- data.frame(
# emb,
# totgexp= totgexp
# )
# 4. clustering
set.seed(10) # for luster labels to remain the same throughout runs
clusters <- as.factor(kmeans(pcs$x[,1:5], centers= 7)$cluster)
# see how many cells are in each cluster
table(clusters)
df <- data.frame(
# emb,
pos,
clusters,
totgexp= totgexp,
# gene=mat[,'Slc34a1'],
pcs$x
)
# highlight cluster of interest in a different colour
# cluster_color_interest <- c(
# "1" = "#D7191C", # red (interest)
# "2" = "#EEEEEE", # light grey
# "3" = "#EEEEEE", # light grey
# "4" = "#EEEEEE", # light grey (is everywhere)
# "5" = "#EEEEEE", # light grey
# "6" = "#EEEEEE", # light grey
# "7" = "#EEEEEE" # light grey (everywhere)
# )
# cluster_colors <- c(
# "1" = "#D6EAF8", # very light blue
# "2" = "#AED6F1",
# "3" = "#5DADE2",
# "4" = "#3498DB",
# "5" = "#2E86C1",
# "6" = "#1B4F72",
# "7" = "#0B3C5D" # deep navy
# )
cluster_colors <- c(
"1" = "#F8766D", # coral
"2" = "#C49A00", # mustard
"3" = "#53B400", # green
"4" = "#00C094", # teal
"5" = "#FB61D7", # sky blue
"6" = "#A58AFF", # lavender
"7" = "#00B6EB" # pink
)
# view clusters
clusters_xenium <- ggplot(df, aes(x=x, y=y, col=clusters))+
geom_point(size=1)+
scale_colour_manual(values = cluster_colors)+
labs(
title= "K means Clusters- Xenium physical space"
)+
theme(plot.title = element_text(size = 11))
clusters_xenium
# view cluster of interest
p_spatial_xenium <- ggplot() +
geom_point(
data = subset(df, clusters != "1"),
aes(x = x, y = y),
color = "#D9D9D9",
alpha = 0.05,
size = 0.6
) +
geom_point(
data = subset(df, clusters == "1"),
aes(x = x, y = y),
color = "#D7191C",
alpha = 0.8,
size = 0.9
) +
labs(
title= "Ascending Loop of Henle- Xenium physical space"
)+
theme_classic() +
theme(
plot.title = element_text(hjust = 0.5, size=11)
)
p_spatial_xenium
# differentially expressed genes in cluster 1
# identify cluster of interest as 1
clusterofinterest_a <- names(clusters)[clusters == 1]
clusterofinterest_b <- names(clusters)[clusters != 1]
# identify differentially expressed genes
out <- sapply(colnames(mat), function(gene){
x1 <- mat[clusterofinterest_a,gene]
x2 <- mat[clusterofinterest_b,gene]
wilcox.test(x1, x2, alternative = "greater")$p.value
})
# the lower the P value, the greater the relative upregulation in x1 compared to x2
sort(out, decreasing = FALSE)[1:30]
# view differentially expressed genes
df <- data.frame(
# emb,
pos,
clusters,
totgexp= totgexp,
mat,
pcs$x
)
plot_slc12a1_xenium <- ggplot(df, aes(x=x, y=y, col=Slc12a1))+geom_point()+
labs(
title= "Scl12a1 Expression- Xenium physical space"
)+
theme(plot.title = element_text(size = 11))
plot_slc12a1_xenium
plot_umod_xenium <- ggplot(df, aes(x=x, y=y, col=Umod))+geom_point()
plot_umod_xenium
# DECONVOLUTION - VISIUM (fixed)
library(STdeconvolve)
# -------------------------
# Load data
# -------------------------
data <- read.csv("~/Documents/genomic-data-visualization-2026/data/Visium-IRI-ShamR_matrix.csv.gz",
stringsAsFactors = FALSE)
pos <- data[,2:3]
colnames(pos) <- c('x', 'y')
cd <- data[, 4:ncol(data)]
rownames(pos) <- rownames(cd) <- data[,1]
## remove pixels with too few genes
counts <- cleanCounts(t(cd), min.lib.size = 100)
## feature select for genes
corpus <- restrictCorpus(counts, removeAbove=1.0, removeBelow = 0.05)
## choose optimal number of cell-types
ldas <- fitLDA(
t(as.matrix(corpus)), # input must be spots x genes
Ks = seq(2, 9, by = 1),
perc.rare.thres = 0.05,
ncores = 8, # adjust to your cluster
verbose = TRUE,
plot = TRUE
)
# ldas <- fitLDA(t(as.matrix(corpus)), Ks = c(5))
## get best model results
# optLDA <- optimalModel(models = ldas, opt = "min")
optLDA <- ldas$models[["6"]]
## extract deconvolved cell-type proportions (theta) and transcriptional profiles (beta)
results <- getBetaTheta(optLDA, perc.filt = 0.05, betaScale = 1000)
deconProp <- results$theta
deconGexp <- results$beta
# # scatterpie
# vizAllTopics(deconProp, pos,
# r=45, lwd=0)
# -------------------------
# scatterbar
# -------------------------
# FIX: align pos to deconProp (cleanCounts removes some spots) + make x/y numeric
pos <- as.data.frame(pos)
pos$x <- as.numeric(pos$x)
pos$y <- as.numeric(pos$y)
pos <- pos[rownames(deconProp), , drop = FALSE]
plot_celltype_proportions <- scatterbar(
deconProp,
pos,
size_x = 80,
size_y = 80,
legend_title = "Cell Types"
)+
labs(title = "STdeconvolve Topic Proportions (K = 6)")+
theme(plot.title = element_text(size = 11))
plot_celltype_proportions
# -------------------------
# k-means on cell-type proportions
# -------------------------
set.seed(10)
theta_mat <- as.matrix(deconProp)
wss <- sapply(2:30, function(k) {
kmeans(theta_mat, centers = k, nstart = 20)$tot.withinss
})
plot(2:30, wss, type = "b",
xlab = "Number of clusters (k)",
ylab = "Total within-cluster sum of squares")
set.seed(10)
k <- 4
kmeans_res <- kmeans(theta_mat, centers = k, nstart = 20)
clusters <- as.factor(kmeans_res$cluster)
names(clusters) <- rownames(theta_mat) # add barcode names (CRUCIAL)
df_cluster <- data.frame(
x = pos$x,
y = pos$y,
cluster = clusters
)
clusters_visium <- ggplot(df_cluster, aes(x = x, y = y, color = cluster)) +
geom_point(size = 2) +
scale_colour_manual(values = cluster_colors)+
labs(
title= "K means Clusters- Visium physical space"
)+
theme(plot.title = element_text(size = 11))
clusters_visium
# -------------------------
# visualize cluster of interest (cluster 3)
# -------------------------
cluster_id <- "3"
p_spatial_visium <- ggplot(df_cluster) +
geom_point(
data = subset(df_cluster, cluster != cluster_id),
aes(x = x, y = y),
color = "#D9D9D9",
alpha = 0.25,
size = 2
) +
geom_point(
data = subset(df_cluster, cluster == cluster_id),
aes(x = x, y = y),
color = "#D7191C",
alpha = 0.9,
size = 2
) +
labs(title = paste0("Ascending Loop of Henle- Visium physical space")) +
theme_classic() +
theme(plot.title = element_text(hjust = 0.5, size=11))
p_spatial_visium
# -------------------------
# differential gene expression: cluster 2 vs all others
# -------------------------
clusterofinterest_a <- names(clusters)[clusters == cluster_id]
clusterofinterest_b <- names(clusters)[clusters != cluster_id]
# clusterofinterest_a <- rownames(deconProp)[5]
# clusterofinterest_b <- rownames(deconProp)[!5]
out <- sapply(colnames(cd), function(gene) {
x1 <- cd[clusterofinterest_a, gene]
x2 <- cd[clusterofinterest_b, gene]
wilcox.test(x1, x2, alternative = "greater")$p.value
})
sort(out, decreasing = FALSE)[1:30]
# view differentially expressed genes
df <- data.frame(
pos,
cd,
clusters
)
plot_slc12a1_visium <- ggplot(df, aes(x=x, y=y, col=Slc12a1))+geom_point(size=3.5)+
labs(
title= "Scl12a1 Expression- Visium physical space"
)+
theme(plot.title = element_text(size = 11))
plot_slc12a1_visium
plot_umod_visium <- ggplot(df, aes(x=x, y=y, col=Umod))+geom_point()
plot_umod_visium
# (plot_celltype_proportions + clusters_xenium + clusters_visum) / (p_spatial_xenium + p_spatial_visium) / (plot_slc12a1_xenium + plot_slc12a1_visium)
# Topic 5 proportion map (aligned to deconProp like scatterbar)
df_t <- data.frame(
pos[rownames(deconProp), , drop = FALSE],
prop = deconProp[, 5]
)
plot_topic_5 <- ggplot(df_t, aes(x = x, y = y, color = prop)) +
geom_point(size = 2) +
theme_void() +
scale_color_viridis_c() +
labs(title = "Topic proportion: 5", color = "Prop")+
theme(plot.title = element_text(size = 11))
plot_topic_5
final_plot <- (plot_celltype_proportions + clusters_xenium + clusters_visium) / (plot_topic_5 + p_spatial_xenium + p_spatial_visium) / (plot_slc12a1_xenium + plot_slc12a1_visium)
final_plot + plot_layout(widths = c(1,1,1))
# save.image("hwEC2.RData")
6. Resources
I used R documentation and the ? help function on R itself to understand functions.
I used AI to help organize plots better