Gene count normalization in single-cell imaging-based spatially resolved transcriptomics
Lyla Atta, Kalen Clifton, Manjari Anant, Gohta Aihara, and Jean Fan^
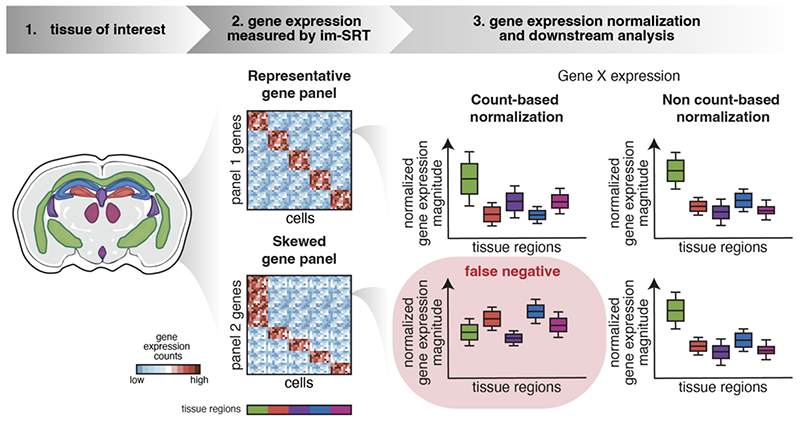
Abstract: Recent advances in imaging-based spatially resolved transcriptomics technologies now enable high-throughput profiling of targeted genes and their locations in fixed tissues. Normalization of gene expression data is often needed to account for technical factors that may confound underlying biological signals. Here, we investigate the potential impact of different gene count normalization methods with different targeted gene panels in the analysis and interpretation of im-SRT data. Using different simulated gene panels that overrepresent genes expressed in specific tissue anatomical regions or cell types, we find that normalization methods that use scaling factors derived from gene counts differentially impact normalized gene expression magnitudes in a region- or cell type-specific manner. We show that these normalization-induced effects may reduce the reliability of downstream differential gene expression and fold change analysis, introducing false positive and false negative results when compared to results obtained from gene panels that are more representative of the gene expression of the tissue's component cell types. These effects are not observed without normalization or when scaling factors are not derived from gene counts, such as with cell volume normalization. Overall, we caution that the choice of normalization method and gene panel may impact the biological interpretation of the im-SRT data.
Paper: Genome Biology. June 12, 2024. doi.org/10.1186/s13059-024-03303-w
| Pubmed
Relevant code: