Most Expressed Genes in Breast Cancer Tissue Single Cell Spatial Transcriptomic Data

What data types are you visualizing?
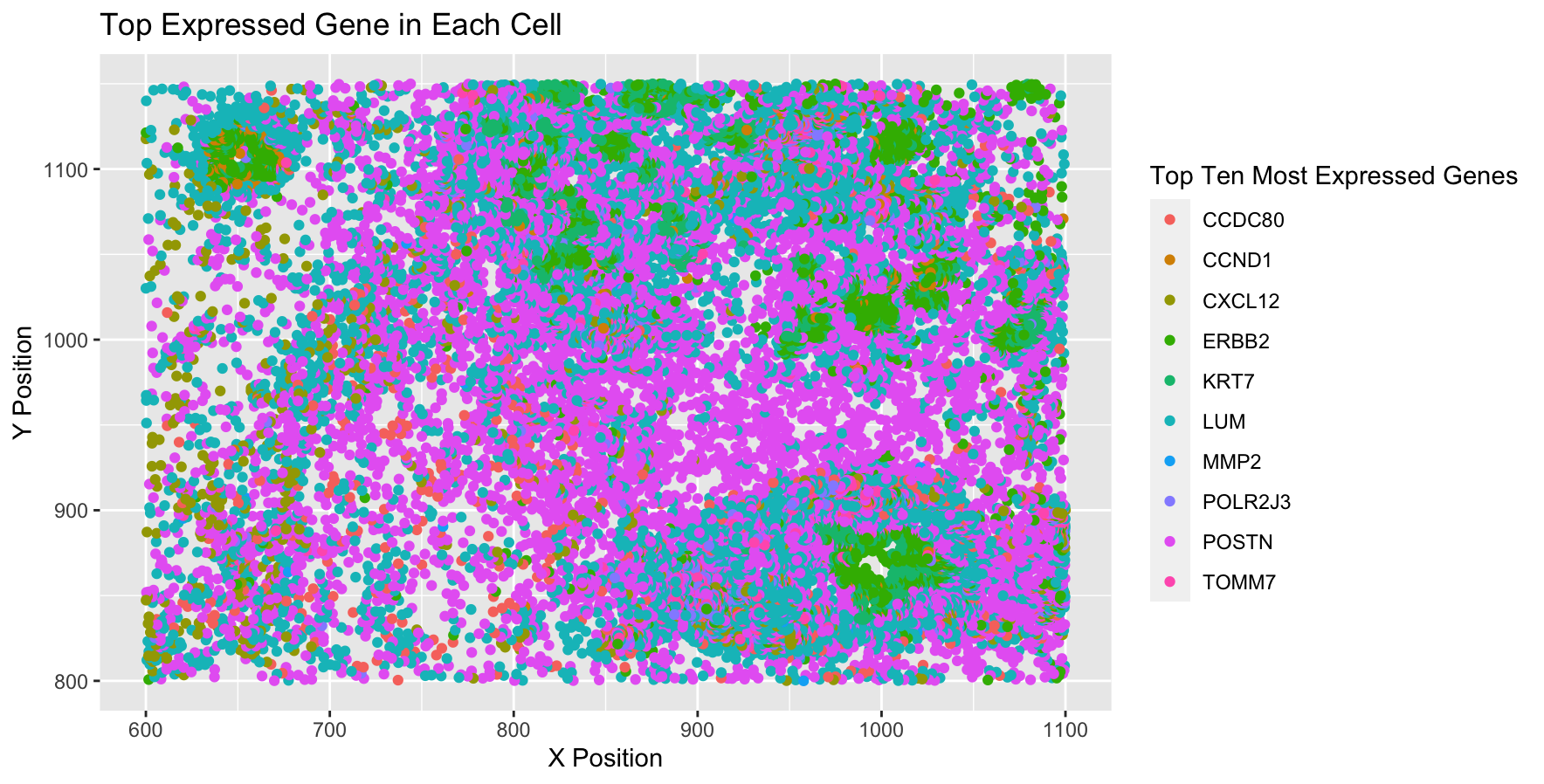
I am visualizing spatial data regarding the x,y centroid positions for each cell, and I am visualizing the categorical data representing the most expressed gene in each cell. Although it is not explicitly calculated, my visualization also shows the quanitative data representing the number of cells with each gene as their most expressed gene.
What data encodings (geometric primitives and visual channels) are you using to visualize these data types?
I am using the geometric primitive of points to represent each cell. To encode the categorical information of which gene is expressed, I am using the visual channel of color. To encode the quantitative information of x,y centroid positions, I am using the visual channel of position. While this isn’t done explicitly, my visualization also uses the geometric primitive of area to represent quantitative data of # of cells expressing each gene as its most expressed gene.
What about the data are you trying to make salient through this data visualization?
My data visualization seeks to make more salient the relationship between gene expression and the cell position. I wanted to highlight how certain genes are broadly expressed while cells expressing other genes are clustered together.
What Gestalt principles or knowledge about perceptiveness of visual encodings are you using to accomplish this?
I used similarity as my main method of demonstrating groups because cells expressing the same genes as their top expressed gene are also the same color. Although I didn’t do this explicitly, the cells expressing the same genes are also grouped by proximity.
data <- read.csv('pikachu.csv.gz', row.names = 1)
library(ggplot2)
library(dplyr)
#find top expressed genes
top_genes <- names(sort(colMeans(data[, 6:318], na.rm = TRUE),
decreasing = TRUE))[1:10]
#filter data to only include columns with top 10 genes
data_filtered <- data %>%
select(aligned_x, aligned_y, all_of(top_genes))
#find the most expressed gene for each cell
filtered_highest_expression <- apply(data_filtered[, 3:12], 1,
function(x) names(x)[which.max(x)])
#highest expressed gene in each cell:
ggplot(data_filtered, aes(x = aligned_x, y = aligned_y,
col = filtered_highest_expression)) +
geom_point() +
labs(title = "Top Expressed Gene in Each Cell",
x = "X Position",
y = "Y Position",
color = "Top Ten Most Expressed Genes")
(I used ChatGPT to assist me in writing the code I used to segment/filter the data and restrict it to most frequently expressed genes)