Identification of a Cluster Associated with Immune Cells

Description of my multi-panel plot
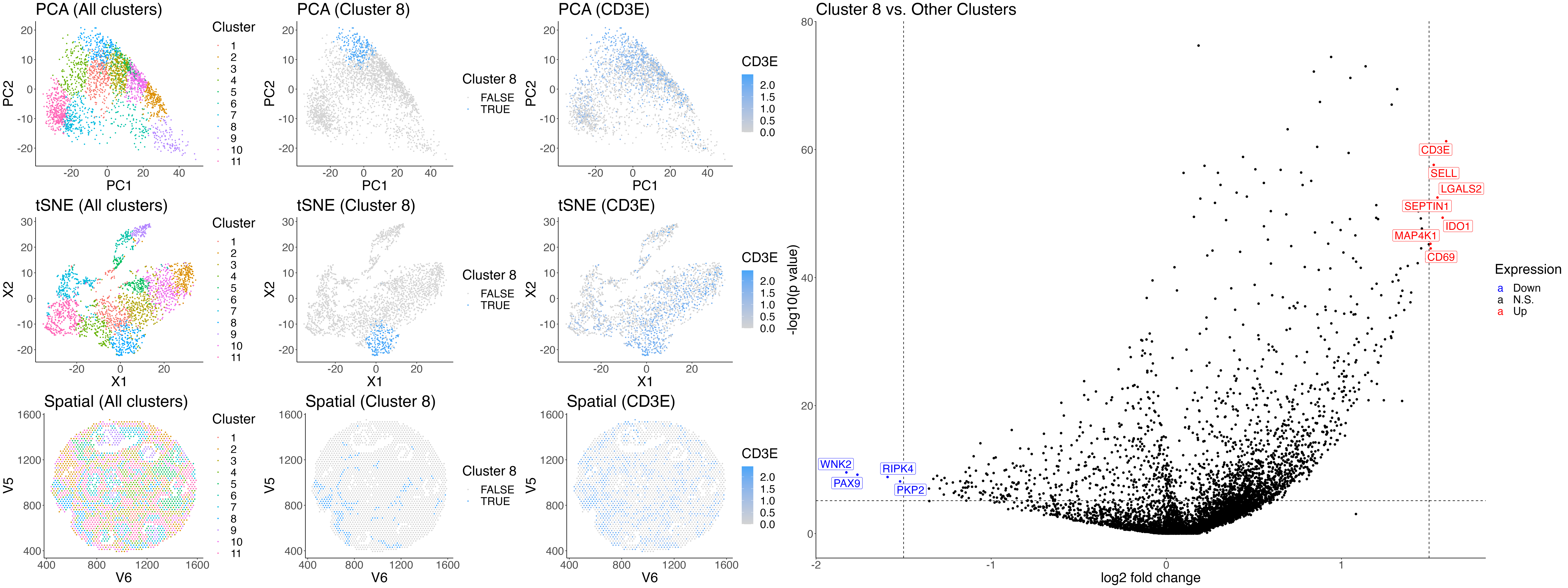
Here, I identified a cluster that seems to include T cells, macrophages, and other immune cells in the Visium breast cancer data. In order to generate the plot above, I normalized the raw gene expression data set by dividing each gene of each spot by the total count of genes for that particular spot. Then, this number is multiplied by 10^6. In other words, the raw gene expression data set was normalized so that each spot’s gene expression represents the count of genes if a spot was to express 10^6 genes. Then, this normalized data set was log10 transformed to create a more Gaussian distribution. Lastly, the data set’s variance was normalized using the scale() function, but mean was left untreated. After normalization and transformation, the data set was analyzed with k means clustering. The number of clusters was determined by plotting WSS and BSS for the number of clusters 1 to 50 and iteratively observing clusters on tSNE plots for different k values. K means clustering was run on the data set with 11. This clustering is used throughout the following analysis. However, it is not certain whether 11 is the optimal cluster number because there was no point in the WSS and BSS plots where the curves started to plateau. The data set was analyzed with PCA and tSNE. In addition, for each cluster, differentially expressed genes were found for that particular cluster against other remaining clusters.
The results were represented on the volcano plot with p value cutoff determined by using the Bonferroni correction and arbitrary determined log2 fold change of 1.5. Volcano plots for each cluster, which represent downregulated genes in blue, upregulated genes in red, and not differentially expressed genes in black, allowed me to identify that cluster 8 most likely contains various immune cells, including T cells and macrophages. As it can be seen from the volcano plot above, differentially expressed genes with highest -log10(p value) and log2 fold change are CD3E, SELL, LGALS2, SEPTIN1, IDO1, MAP4K1, CD69. CD3E encodes for CD3 epsilon subunit of T cell receptor complex, and it is commonly used for T cell markers [1]. SELL encodes for L Selectin (also known as CD62L), which is a type of cell surface adhesion molecules [2]. According to the literature, CD62L is expressed by all leukocytes, such as neutrophils, T cells, and monocytes, except for activated, memory lymphocytes [3]. Further, CD62L us known as a “homing receptor” because it facilitates a process called “leukocyte rolling” and allow leukocytes to enter inflammatory sites from the blood vessel [3]. LGALS2 encodes for another type of lectin called Galectin 2 with specificity for beta-galactose [4]. Functions of Galectin 2 are not well understood yet, but their observed effects are associated with immune cells. For example, Galectin 2 can induce T cell apoptosis, and it also skews macrophages into anti-inflammatory M2 phenotype [5,6]. SEPTIN1 encodes for Septin 1, which is a type of GTPases required for cytokinesis and the maintenance of cell morphology [7]. The Human Protein Atlas has shown that SEPTIN1 is highly expressed in immune cells, especially in T cells [8,9]. IDO1 encodes for indoleamine 2,3-dioxygenase 1 (IDO1), which is a type of enzyme that plays a role in tryptophan catabolism [10]. Studies have shown that IDO1 is widely expressed in both immune and non-immune cells, and it seems to inhibit T cell activation by increasing the expression of immune checkpoint molecule, PD-1, in T cells [11,12]. MAP4K1 encodes for Mitogen-Activated Protein Kinase Kinase Kinase Kinase 1 [13]. MAP4K1 works with other MAP kinase pathway proteins to facilitate signal transductions in various pathways [14]. Interestingly, MAP4K1 is usually highly expressed by myeloid and lymphoid lineage cells, such as monocytes and T cells. Lastly, CD69 encodes for CD69 surface receptors, which are known to be induced upon T cell activation [15]. Based on these information, some upregulated genes in cluster 8 are specifically associated with T cells, monocytes, or macrophages, while other upregulated genes are associated with all immune cells. Therefore, it can be concluded that cluster 8 most likely contains various immune cells, including T cells and macrophages. Rather ambiguous identification of a cell type that is associated with cluster 8 is most likely due to the inherent property of Visium itself. Unlike MERFISH data set, it is important to note that Visium is not a single cell-based technology. Since each spot can capture RNA molecules from multiple cells that ended up on the spot, co-localization of different cell types most likely result in a spot with differentially expressed genes associated with multiple types of cells. Given that the data set is from a breast cancer section, it can be hypothesized that various immune cells co-localized in the tumor microenvironment. This ambiguity can be observed in PCA, tSNE, and spatial plots for CD3E. Although CD3E expression fairly matches with the location of cluster 8 in each types of plots, there are other clusters’ spots that show high expression of CD3E. These spots might have contained RNA molecules from T cells and other non-immune cells, such as breast cancer cells, such that despite high level of CD3E expression, their transcriptomic profiles became far enough for them to be categorized as other clusters.
References
- https://www.ncbi.nlm.nih.gov/gene/916
- https://www.genecards.org/cgi-bin/carddisp.pl?gene=SELL
- https://www.sciencedirect.com/topics/neuroscience/l-selectin
- https://www.genecards.org/cgi-bin/carddisp.pl?gene=LGALS2
- https://academic.oup.com/abbs/article/48/10/939/2389061
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9242595/#:~:text=The%20previous%20literature%20suggests%20that,monocytes%20and%20macrophages%20(15).
- https://www.genecards.org/cgi-bin/carddisp.pl?gene=SEPTIN1
- https://www.proteinatlas.org/ENSG00000180096-SEPTIN1/immune+cell
- https://www.proteinatlas.org/ENSG00000180096-SEPTIN1
- https://www.ncbi.nlm.nih.gov/gene/3620
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8136272/
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7461966/#:~:text=IDO1%20has%20been%20shown%20to,evasion%20(13%E2%80%9315).
- https://www.ncbi.nlm.nih.gov/gene/11184
- https://www.nature.com/articles/7290105#:~:text=MAPK%20pathways%20relay%2C%20amplify%20and,and%20apoptosis%20in%20mammalian%20cells.
- https://www.ncbi.nlm.nih.gov/gene/969
Code
setwd('~/Desktop/Johns Hopkins University/4 - Senior (2022 - 2023)/Spring/Genomic Data Visualization/HW/HW6')
getwd()
## load library
library(ggplot2)
library(Rtsne)
library(gridExtra)
library(ggrepel)
## set seed for reproducibility
set.seed(0)
## set 1x1 plot space
par(mfrow=c(1,1))
## read in data
data <- read.csv('~/Desktop/genomic data visualization 2023 data (copy)/visium_breast_cancer.csv.gz', row.names = 1)
gexp <- data[,3:ncol(data)]
dim(gexp) # 2518 cells x 7037 genes
genes <- colnames(gexp)
pos <- data[,1:2]
#### data exploration ####
## QC
hist(log10(rowSums(gexp+1)))
hist(log10(gexp[,'NOC2L']+1))
## normalize
mat <- gexp/rowSums(gexp) * 1e+6
mat <- log10(mat + 1)
## check variance
head(sort(apply(mat, 2, var), decreasing = TRUE))
head(sort(apply(mat, 2, var), decreasing = FALSE))
hist(apply(mat, 2, var))
## scale
mat <- scale(mat, center=FALSE, scale = TRUE)
## PCA
set.seed(0)
pcs <- prcomp(mat)
plot(1:40, pcs$sdev[1:40], type='b',main='PCA STDEV')
## kmeans clustering
## characterize kmeans clustering
## find optimal number of k
ks <- 1:30
## loop through ks to return wss and bss as matrix (only use first 30 PCs for faster computation)
out <- do.call(rbind, lapply(ks, function(k) {
com <- kmeans(pcs$x[,1:30], centers = k)
c(within = com$tot.withinss, between = com$betweenss)
}))
plot(ks,out[,1],type='b',main='WSS')
plot(ks,out[,2],type='b',main='BSS')
## kmeans clustering
num_center = 11
set.seed(0)
com <- kmeans(mat, centers=num_center)
## tSNE
set.seed(0)
emb <- Rtsne(mat)
# plot tSNE analysis results
df <- data.frame(pos, emb$Y, celltype = as.factor(com$cluster))
head(df)
#ggplot(df, aes(x = X1, y = X2)) + geom_point() + theme_classic()
p1 <- ggplot(df, aes(x = V6, y = V5, col=celltype)) +
geom_point(size = 0.3) +
theme_classic()
p2 <- ggplot(df, aes(x = X1, y = X2, col=celltype)) +
geom_point(size = 0.3) +
theme_classic()
grid.arrange(p1,p2)
## differential gene expression
## loop through each cluster to find differentially expressed genes for each cluster vs. remaining clusters
volcano_plt <- lapply(seq_len(num_center), function(i) {
## pick a cluster
cluster.of.interest <- names(which(com$cluster == i))
cluster.other <- names(which(com$cluster != i))
## loop through my genes and test each one
out <- sapply(genes, function(g) {
a <- mat[cluster.of.interest, g]
b <- mat[cluster.other, g]
pvs <- wilcox.test(a,b,alternative='two.sided')$p.val
#pvs <- as.data.frame(pvs)
log2fc <- log2(mean(a)/mean(b))
#log2fc <- as.data.frame(log2fc)
c(pvs=pvs,log2fc=log2fc)
})
## volcano plot
# run Bonferroni correction determine a p value cutoff (conservative method with low false-positive rate but high false-negative rate as a trade-off)
cutoff.pvs <- 0.05/ncol(mat) # false-positive rate would be 1-(1-0.05/ncol(mat))^ncol(mat)
cutoff.log2fc <- 1.5
df <- data.frame(pvs=out[1,], log2fc=out[2,])
## prepare a data frame for plotting
df2 <- df
# add a column of NAs to df2 titled diffexpressed
df2$diffexpressed <- 'NO'
# if log2fc > cutoff.log2fc and pvalue < cutoff.pvs, set as "Up"
df2$diffexpressed[df2$log2fc > cutoff.log2fc & df2$pvs < cutoff.pvs] <- "Up"
# if log2fc < -cutoff.log2fc and pvalue < cutoff.pvs, set as "Down"
df2$diffexpressed[df2$log2fc < -cutoff.log2fc & df2$pvs < cutoff.pvs] <- "Down"
# add a column of NAs to df2 titled genelabel that will contain names of differentially expressed genes
df2$genelabel <- NA
df2$genelabel[df2$diffexpressed != 'NO'] <- rownames(df2)[df2$diffexpressed != 'NO']
plt <- ggplot(df2, aes(x=log2fc,y=-log10(pvs), col=diffexpressed, label=genelabel)) +
geom_point() +
scale_color_manual(values = c('blue', 'black', 'red'),
name = 'Expression',
breaks = c('Down', 'NO', 'Up'),
labels = c('Down', 'N.S.', 'Up')) +
ggtitle(paste('Cluster',i,'vs. Other Clusters')) +
xlab('log2 fold change') +
ylab('-log10(p value)') +
#geom_label_repel() +
geom_vline(xintercept = c(-cutoff.log2fc, cutoff.log2fc), col='black',linetype='dashed') +
geom_hline(yintercept = -log10(cutoff.pvs), col='black',linetype='dashed') +
theme_classic()
plt
})
## save all figures
for (i in seq_len(num_center)) {
name = paste0('volcano_cluster_',num_center,'clusters_',i,'_vs_otherclusters.png')
plt <- volcano_plt[[i]]
ggsave(name, plot = plt, width = 10, height = 10, dpi = 300)
}
#### plot for a figure ####
## set custom theme for plots
plot_theme <- theme_classic() +
theme(
text = element_text(size = 25),
legend.key.size = unit(0.75, 'cm')
)
## plot certain cluster on tSNE and spatial
cluster.of.interest = 8
gene.of.interest = 'CD3E'
df <- data.frame(pos, pcs$x[,1:2], emb$Y, celltype = as.factor(com$cluster), gene=mat[,gene.of.interest])
head(df)
# PCA plots
p1 <- ggplot(df, aes(x = PC1, y = PC2, col = celltype)) +
geom_point(size = 0.5) +
ggtitle('PCA (All clusters)') +
labs(color = 'Cluster') +
plot_theme
p2 <- ggplot(df, aes(x = PC1, y = PC2, col = celltype == cluster.of.interest)) +
geom_point(size = 0.5) +
scale_color_manual(values = c('lightgrey','#46A5F8'),
name = paste('Cluster',cluster.of.interest),
labels = c('FALSE','TRUE')) +
ggtitle(paste0('PCA (Cluster ',cluster.of.interest,')')) +
plot_theme
p3 <- ggplot(df, aes(x = PC1, y = PC2, col = gene)) +
geom_point(size = 0.5) +
scale_color_gradient(low='lightgrey', high='#46A5F8', name = gene.of.interest) +
ggtitle(paste0('PCA (',gene.of.interest,')')) +
plot_theme
# tSNE plots
p4 <- ggplot(df, aes(x = X1, y = X2, col = celltype)) +
geom_point(size = 0.5) +
ggtitle('tSNE (All clusters)') +
labs(color = 'Cluster') +
plot_theme
p5 <- ggplot(df, aes(x = X1, y = X2, col = celltype == cluster.of.interest)) +
geom_point(size = 0.5) +
scale_color_manual(values = c('lightgrey','#46A5F8'),
name = paste('Cluster',cluster.of.interest),
labels = c('FALSE','TRUE')) +
ggtitle(paste0('tSNE (Cluster ',cluster.of.interest,')')) +
plot_theme
p6 <- ggplot(df, aes(x = X1, y = X2, col = gene)) +
geom_point(size = 0.5) +
scale_color_gradient(low='lightgrey', high='#46A5F8', name = gene.of.interest) +
ggtitle(paste0('tSNE (',gene.of.interest,')')) +
plot_theme
# spatial plots
p7 <- ggplot(df, aes(x = V6, y = V5, col = celltype)) +
geom_point(size = 0.5) +
ggtitle('Spatial (All clusters)') +
labs(color = 'Cluster') +
plot_theme
p8 <- ggplot(df, aes(x = V6, y = V5, col = celltype == cluster.of.interest)) +
geom_point(size = 0.5) +
scale_color_manual(values = c('lightgrey','#46A5F8'),
name = paste('Cluster',cluster.of.interest),
labels = c('FALSE','TRUE')) +
ggtitle(paste0('Spatial (Cluster ',cluster.of.interest,')')) +
plot_theme
p9 <- ggplot(df, aes(x = V6, y = V5, col = gene)) +
geom_point(size = 0.5) +
scale_color_gradient(low='lightgrey', high='#46A5F8', name = gene.of.interest) +
ggtitle(paste0('Spatial (',gene.of.interest,')')) +
plot_theme
## volcano plot
p10 <- volcano_plt[[cluster.of.interest]] +
geom_label_repel(size = 7) +
plot_theme
lay <- rbind(c(1,2,3,10,10,10),
c(4,5,6,10,10,10),
c(7,8,9,10,10,10))
result <- grid.arrange(p1, p2, p3, p4, p5, p6, p7, p8, p9, p10, layout_matrix = lay)
## save plot
ggsave("hw6_gaihara1.png", plot = result, width = 40, height = 15, dpi = 300)
#### references ####
# information on sapply(): https://www.guru99.com/r-apply-sapply-tapply.html
# return multiple output for apply(): https://stackoverflow.com/questions/12300565/return-multiple-values-from-apply-function-in-r
# volcano plots: https://biocorecrg.github.io/CRG_RIntroduction/volcano-plots.html
# linetype: http://sape.inf.usi.ch/quick-reference/ggplot2/linetype
# bonferroni correction: https://www.aaos.org/aaosnow/2012/apr/research/research7/#:~:text=The%20Bonferroni%20correction%20is%20an,number%20of%20comparisons%20being%20made.
# bonferroni correction 2: https://www.youtube.com/watch?v=HLzS5wPqWR0&ab_channel=TopTipBio
# ggrepel package (in Japanese): https://id.fnshr.info/2017/03/19/ggrepel/
# most of the codes were taken from HW5
r#:~:text=Example%201%3A%20Remove%20All%20Legends%20in%20ggplot2&text=We%20simply%20had%20to%20specify,get%20rid%20of%20both%20legends.
(do you think my data visualization is effective? why or why not?)