EC

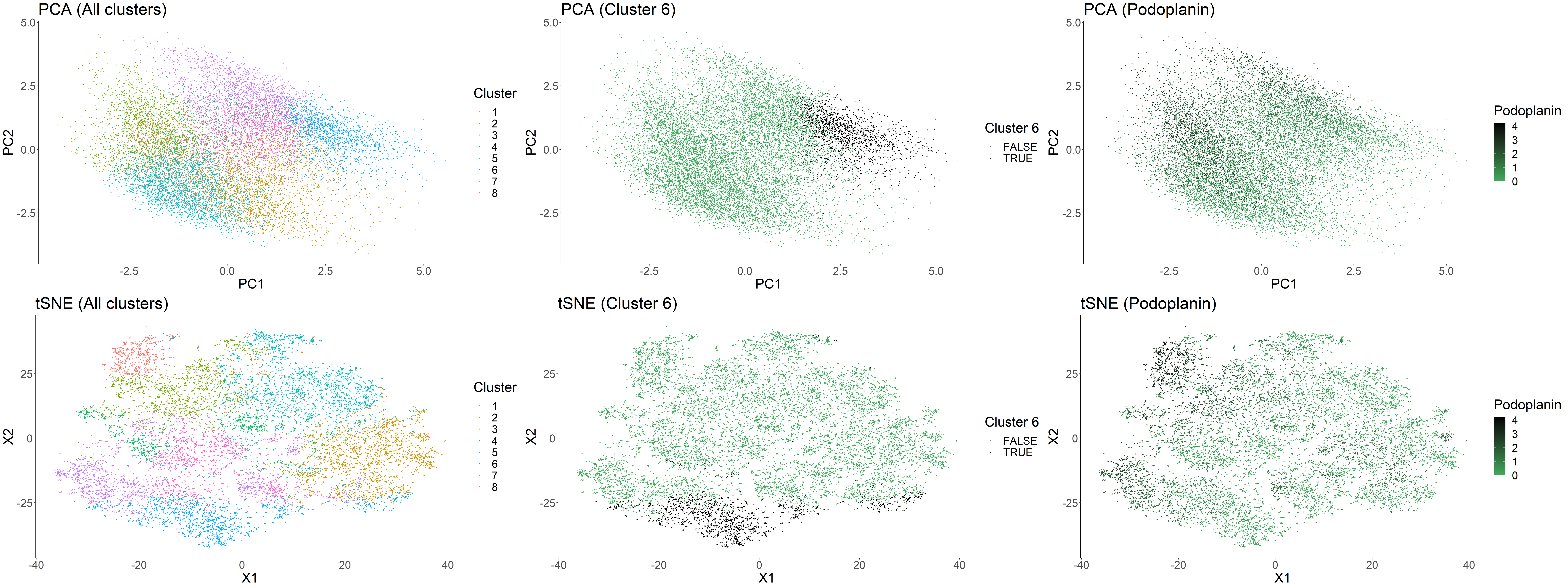
Comparing the gene expressions between clusters revealed that cluster 6 had a higher expression value for Podoplanin. To determine the optimal cluster, the highest gene expression value for a specific gene was compared across all clusters, with the cluster having the highest expression level being selected. In this case, cluster 6 had the highest expression level for Podoplanin, with a value of 7.90E-290, making it the most relevant cluster for this gene. Therefore, cluster 6 is considered the best cluster to choose from. Based on the significance of the gene expressed Podoplanin, the cell type to which this cluster belongs would most likely be lymphatic endothelial cells, but it can also correspond to follicular dendritic cells, mesothelial cells, and type I alveolar cells in the lung.
Reference: https://www.ncbi.nlm.nih.gov/gene/10630
code
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
## load library
library(ggplot2)
library(Rtsne)
library(gridExtra)
library(ggrepel)
## set seed for reproducibility
set.seed(0)
## set 1x1 plot space
par(mfrow=c(1,1))
############################
# Quality Control
############################
## read in data
data <- read.csv('codex_spleen_subset.csv', row.names = 1)
gexp <- data[,4:ncol(data)]
genes <- colnames(gexp)
pos <- data[,1:2]
# normalize
totgexp <- rowSums(gexp)
mat <- gexp/rowSums(gexp) * median(rowSums(gexp))
mat <- log10(mat + 1)
############################
# Kmeans clustering
############################
# find optimal number of k
library(segmented)
ks <- 1:28
# loop through ks to return wss and bss as matrix
out <- do.call(rbind, lapply(ks, function(k) {
com <- kmeans(mat, centers = k)
c(within = com$tot.withinss, between = com$betweenss)
}))
fit <- segmented(lm(out[,1] ~ ks))
breakpoint <- fit$psi[1]
plot(ks, out[,1], type='b', main='WSS')
plot(ks,out[,2],type='b',main='BSS')
abline(fit, col='red')
abline(v=breakpoint, col='blue')
# optimal number of k=10
num_center = 8
com <- kmeans(mat, centers=num_center)
############################
# Dimensionality Reduction
############################
## PCA analysis
pcs <- prcomp(mat)
## tSNE analysis
emb <- Rtsne(mat)
# plot tSNE analysis results
df <- data.frame(pos, emb$Y, pcs$x, celltype = as.factor(com$cluster))
head(df)
p1 <- ggplot(df, aes(x = X1, y = X2, col=celltype)) +
geom_point(size = 0.01) +
scale_color_manual(values=c("#1b9e77", "#d95f02", "#7570b3", "#e7298a", "#66a61e", "#e6ab02", "#a6761d", "#666666", "#1f78b4", "#b2df8a", "#33a02c", "#fb9a99", "#e31a1c", "#fdbf6f", "#ff7f00")) +
theme_classic()
p2 <- ggplot(df, aes(x = PC1, y = PC2, col=celltype)) +
geom_point(size = 0.01) +
scale_color_manual(values=c("#1b9e77", "#d95f02", "#7570b3", "#e7298a", "#66a61e", "#e6ab02", "#a6761d", "#666666", "#1f78b4", "#b2df8a", "#33a02c", "#fb9a99", "#e31a1c", "#fdbf6f", "#ff7f00")) +
theme_classic()
grid.arrange(p1,p2)
ggplot(df, aes(x=x, y=y, col=celltype)) +
geom_point(size=0.01)
############################
# Pick one cluster
############################
## pick a cluster
cluster_number = 6
cluster.of.interest <- names(which(com$cluster == cluster_number))
cluster.other <- names(which(com$cluster != cluster_number))
## loop through my genes and test each one
out <- sapply(genes, function(g) {
a <- mat[cluster.of.interest, g]
b <- mat[cluster.other, g]
pvs <- wilcox.test(a,b,alternative='two.sided')$p.val
#pvs <- as.data.frame(pvs)
log2fc <- log2(mean(a)/mean(b))
#log2fc <- as.data.frame(log2fc)
c(pvs=pvs,log2fc=log2fc)
})
## volcano plot
# run Bonferroni correction determine a p value cutoff (conservative method with low false-positive rate but high false-negative rate as a trade-off)
cutoff.pvs <- 0.05/ncol(mat) # false-positive rate would be 1-(1-0.05/ncol(mat))^ncol(mat)
cutoff.log2fc <- 1
df_temp <- data.frame(pvs=out[1,], log2fc=out[2,])
## prepare a data frame for plotting
df2 <- df_temp
# add a column of NAs to df2 titled diffexpressed
df2$diffexpressed <- 'NO'
# if log2fc > cutoff.log2fc and pvalue < cutoff.pvs, set as "Up"
df2$diffexpressed[df2$log2fc > cutoff.log2fc & df2$pvs < cutoff.pvs] <- "Up"
# if log2fc < -cutoff.log2fc and pvalue < cutoff.pvs, set as "Down"
df2$diffexpressed[df2$log2fc < -cutoff.log2fc & df2$pvs < cutoff.pvs] <- "Down"
# add a column of NAs to df2 titled genelabel that will contain names of differentially expressed genes
df2$genelabel <- NA
df2$genelabel[df2$diffexpressed != 'NO'] <- rownames(df2)[df2$diffexpressed != 'NO']
## get the most significant genes (upregulated) from the chosen cluster
significant_genes <- df2$gene[df2$diffexpressed == 'Up']
#sort by p-value
significant_genes_sorted <- significant_genes[order(out[1,][significant_genes])]
#choose first one from list
most_significant_gene <- significant_genes_sorted[1]
volcano_plt <- ggplot(df2, aes(x=log2fc,y=-log10(pvs), col=diffexpressed, label=genelabel)) +
geom_point() +
scale_color_manual(values = c('blue', 'black', 'red'),
name = 'Expression',
breaks = c('Down', 'NO', 'Up'),
labels = c('Down', 'N.S.', 'Up')) +
ggtitle(paste('Cluster ',cluster_number,' vs. Other Clusters')) +
xlab('log2 fold change') +
ylab('-log10(p value)') +
geom_label_repel() +
geom_vline(xintercept = c(-cutoff.log2fc, cutoff.log2fc), col='black',linetype='dashed') +
geom_hline(yintercept = -log10(cutoff.pvs), col='black',linetype='dashed') +
theme_classic()
volcano_plt
significant_genes
most_significant_gene
############################
# Multi-Panel Visualization
############################
#### plot for a fi
gure ####
## set custom theme for plots
plot_theme <- theme_classic() +
theme(
text = element_text(size = 25),
legend.key.size = unit(0.75, 'cm')
)
# set the green color palette
green_palette <- c("#e5f5e0", "#c7e9c0", "#a1d99b", "#74c476", "#41ab5d", "#238b45", "#006d2c", "#00441b")
# plot certain cluster on tSNE
cluster.of.interest = cluster_number
gene.of.interest = most_significant_gene
df <- data.frame(pos, pcs$x[,1:2], emb$Y, celltype = as.factor(com$cluster), gene=mat[,gene.of.interest])
head(df)
# PCA plots
p1 <- ggplot(df, aes(x = PC1, y = PC2, col = celltype)) +
geom_point(size = 0.5) +
ggtitle('PCA (All clusters)') +
labs(color = 'Cluster') +
plot_theme
p2 <- ggplot(df, aes(x = PC1, y = PC2, col = celltype == cluster.of.interest)) +
geom_point(size = 0.5) +
scale_color_manual(values = c(green_palette[5], 'black'),
name = paste('Cluster',cluster.of.interest),
labels = c('FALSE','TRUE')) +
ggtitle(paste0('PCA (Cluster ',cluster.of.interest,')')) +
plot_theme
p3 <- ggplot(df, aes(x = PC1, y = PC2, col = gene)) +
geom_point(size = 0.5) +
scale_color_gradient(low=green_palette[5], high='black', name = gene.of.interest) +
ggtitle(paste0('PCA (',gene.of.interest,')')) +
plot_theme
# tSNE plots
p4 <- ggplot(df, aes(x = X1, y = X2, col = celltype)) +
geom_point(size = 0.5) +
ggtitle('tSNE (All clusters)') +
labs(color = 'Cluster') +
plot_theme
p5 <- ggplot(df, aes(x = X1, y = X2, col = celltype == cluster.of.interest)) +
geom_point(size = 0.5) +
scale_color_manual(values = c(green_palette[5], 'black'),
name = paste('Cluster',cluster.of.interest),
labels = c('FALSE','TRUE')) +
ggtitle(paste0('tSNE (Cluster ',cluster.of.interest,')')) +
plot_theme
p6 <- ggplot(df, aes(x = X1, y = X2, col = gene)) +
geom_point(size = 0.5) +
scale_color_gradient(low=green_palette[5], high='black', name = gene.of.interest) +
ggtitle(paste0('tSNE (',gene.of.interest,')')) +
plot_theme
lay <- rbind(c(1,2,3),
c(4,5,6))
plot_theme <- plot_theme + theme(plot.width = unit(5, "cm"), plot.height = unit(5, "cm"))
result <- grid.arrange(p1, p2, p3, p4, p5, p6, layout_matrix = lay)
## save panel plots
ggsave("hw_moneera_panels.png", plot = result, width = 40, height = 15, dpi = 300)