Identification of a Cluster Associated with CD8+ T cells

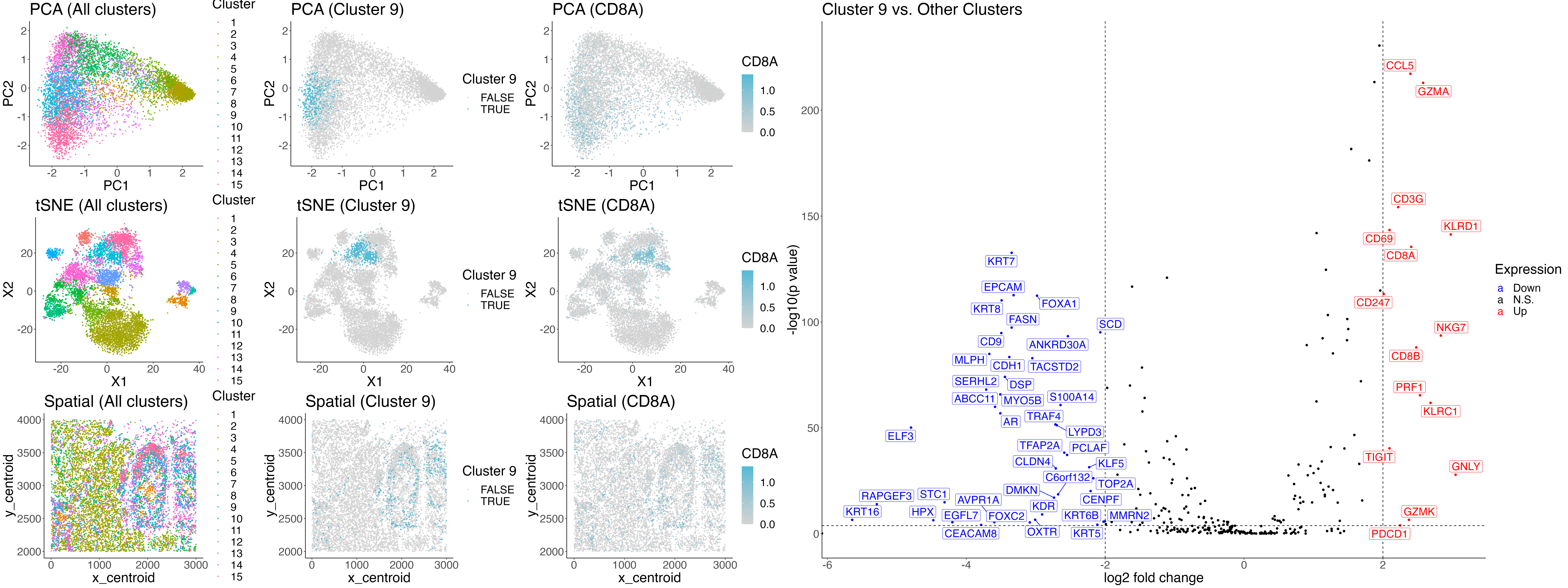
Description of my multi-panel plot
Here, I identified a cluster that seems to be CD8+ T cells. In order to generate the plot above, I normalized the raw gene expression data set by dividing each gene of each cell by the total count of genes for that particular cell. Then, this number is multiplied by the median value of the total count of genes for all cells. In other words, the raw gene expression data set was normalized so that each cell’s gene expression represents the count of genes if a cell was to express median value of the total gene count. Then, this normalized data set was log10 transformed to create a more Gaussian distribution. After normalization and transformation, the data set was analyzed with k means clustering. The number of clusters was determined by plotting WSS and BSS for the number of clusters 1 to 50. K means clustering was run on the data set with 15 as the number of clusters as it was the point when both WSS and BSS curves seemed to plateau. This clustering is used throughout the following analysis. The data set was analyzed with PCA and tSNE. In addition, for each cluster, differentially expressed genes were found for that particular cluster against other remaining clusters. The results of this were represented on the volcano plot with p value cutoff determined by using the Bonferroni correction and arbitrary determined log2 fold change of 2. Volcano plots for each cluster, which represent downregulated genes in blue, upregulated genes in red, and not differentially expressed genes in black, allowed me to identify that cluster 9 is most likely CD8+ T cells. As it can be seen from the volcano plot above, differentially expressed genes with highest -log10(p value) and log2 fold change are CCL5, GZMA, CD3G, CD69, KLRD1, CD8A, CD8B, PRF1, and NKG7. CD8A and CD8B encode for CD8 alpha and CD8 beta surface proteins, which are commonly used markers for CD8+ T cells, especially in flow cytometry [1, 2]. In addition, CD3G encodes for gamma subunit for T cell receptor complex, which is also regularly expressed in CD8+ T cells [3]. CD69 encodes for CD69 surface receptors, which are known to be induced upon T cell activation [4]. GZMA and PRF1 encodes for cytotoxic molecule called granzyme A and perforin 1, respectively, which are both released by cytotoxic (CD8+) T cells and NK cells to lyse their targets [5, 6]. CCL5 encodes for chemokine ligand 5 (CCL5). Similar to other chemokine molecules, CCL5 functions as chemoattractant for blood monocytes, memory T cells, and eosinophils [7]. However, a study has shown that high level of CCL5 is expressed by CD8+ effector T cells, which are a subset of CD8+ T cells that mediate direct killing of targets, such as tumor cells [8]. It is interesting to note that cluster 9 also has upregulated KLRD1 and NKG7, which are both often associated with NK cells [9, 10]. Nonetheless, studies have indicated that KLRG1 can be expressed during CD8+ T cell differentiation, and NKG7 mRNA is expressed in activated T cells [11, 12]. These findings indicate that cluster 9 is most likely be a mixture of CD8+ effector T cells with cytolytic abilities and CD8+ T cells that are activated and differentiating towards CD8+ effector T cells. In addition, the volcano plot shows that one of the identified downregulated genes in cluster 9 is EPCAM. EPCAM encodes for epithelial cellular adhesion molecule (EpCAM), which is expressed by epithelial cells [12]. Since CD8+ T cells are not epithelial cells, downexpression of EPCAM corresponds with the hypothesis that cluster 9 is most likely CD8+ T cells. It is important to note that our analysis on the identity of cluster 9 is not perfect. As it can be seen from PCA, tSNE, and spatial plots, high expression of CD8A, a commonly used marker for CD8+ T cells, does not completely match cluster 9. Rather, half of cluster 9 express high level of CD8A, while a portion of cluster 14 also express high level of CD8A. This can be due to the number of clusters set for k means clustering. It is possible that since I qualitatively picked the number of clusters from WSS and BSS plots, the optimal number of clusters is actually lower or higher. If the optimal number of clusters is lower, a cluster that is equivalent to CD8+ T cells got further divided into cluster 9 and cluster 14. On the other hand, if the optimal number of clusters is higher, cluster 9 can possibly be further divided into two clusters with low expression of CD8A and high expression of CD8A. In this case, a portion of cluster 9 with high expression of CD8A would represent CD8+ T cells.
References
- https://www.genecards.org/cgi-bin/carddisp.pl?gene=CD8A
- https://www.genecards.org/cgi-bin/carddisp.pl?gene=CD8B
- https://www.ncbi.nlm.nih.gov/gene/917
- https://www.ncbi.nlm.nih.gov/gene/969
- https://www.ncbi.nlm.nih.gov/gene/3001
- https://www.genecards.org/cgi-bin/carddisp.pl?gene=PRF1
- https://www.ncbi.nlm.nih.gov/gene/6352
- https://www.frontiersin.org/articles/10.3389/fimmu.2022.887972/full#:~:text=CD8%20Effector%20T%20Cells%2C%20in,as%20Compared%20to%20Healthy%20Controls
- https://www.genecards.org/cgi-bin/carddisp.pl?gene=KLRD1
- https://www.genecards.org/cgi-bin/carddisp.pl?gene=NKG7
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2262179/
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1934518/#:~:text=Expression%20of%20EpCAM%20in%20Mature,for%20tissue%20maintenance%20and%20turnover.
Code
setwd('~/Desktop/Johns Hopkins University/4 - Senior (2022 - 2023)/Spring/Genomic Data Visualization/HW/HW5')
getwd()
## load library
library(ggplot2)
library(Rtsne)
library(gridExtra)
library(ggrepel)
## set seed for reproducibility
set.seed(0)
## set 1x1 plot space
par(mfrow=c(1,1))
## read in data
data <- read.csv('~/Desktop/genomic data visualization 2023 data (copy)/bulbasaur.csv.gz', row.names = 1)
gexp <- data[,4:ncol(data)]
genes <- colnames(gexp)
pos <- data[,1:2]
#### data exploration ####
# normalize
totgexp <- rowSums(gexp)
mat <- gexp/rowSums(gexp) * median(rowSums(gexp))
mat <- log10(mat + 1)
## kmeans clustering
# characterize kmeans clustering
# find optimal number of k
ks <- 1:50
# loop through ks to return wss and bss as matrix
out <- do.call(rbind, lapply(ks, function(k) {
com <- kmeans(mat, centers = k)
c(within = com$tot.withinss, between = com$betweenss)
}))
plot(ks,out[,1],type='b',main='WSS')
plot(ks,out[,2],type='b',main='BSS')
# kmeans clustering
num_center = 15
com <- kmeans(mat, centers=num_center)
## PCA analysis
pcs <- prcomp(mat)
## tSNE analysis
emb <- Rtsne(mat)
# plot tSNE analysis results
df <- data.frame(pos, emb$Y, celltype = as.factor(com$cluster))
head(df)
p1 <- ggplot(df, aes(x = x_centroid, y = y_centroid, col=celltype)) +
geom_point(size = 0.3) +
theme_classic()
p2 <- ggplot(df, aes(x = X1, y = X2, col=celltype)) +
geom_point(size = 0.3) +
theme_classic()
grid.arrange(p1,p2)
## loop through each cluster to find differentially expressed genes for each cluster vs. remaining clusters
volcano_plt <- lapply(seq_len(num_center), function(i) {
## pick a cluster
cluster.of.interest <- names(which(com$cluster == i))
cluster.other <- names(which(com$cluster != i))
## loop through my genes and test each one
out <- sapply(genes, function(g) {
a <- mat[cluster.of.interest, g]
b <- mat[cluster.other, g]
pvs <- wilcox.test(a,b,alternative='two.sided')$p.val
#pvs <- as.data.frame(pvs)
log2fc <- log2(mean(a)/mean(b))
#log2fc <- as.data.frame(log2fc)
c(pvs=pvs,log2fc=log2fc)
})
## volcano plot
# run Bonferroni correction determine a p value cutoff (conservative method with low false-positive rate but high false-negative rate as a trade-off)
cutoff.pvs <- 0.05/ncol(mat) # false-positive rate would be 1-(1-0.05/ncol(mat))^ncol(mat)
cutoff.log2fc <- 2
df <- data.frame(pvs=out[1,], log2fc=out[2,])
## prepare a data frame for plotting
df2 <- df
# add a column of NAs to df2 titled diffexpressed
df2$diffexpressed <- 'NO'
# if log2fc > cutoff.log2fc and pvalue < cutoff.pvs, set as "Up"
df2$diffexpressed[df2$log2fc > cutoff.log2fc & df2$pvs < cutoff.pvs] <- "Up"
# if log2fc < -cutoff.log2fc and pvalue < cutoff.pvs, set as "Down"
df2$diffexpressed[df2$log2fc < -cutoff.log2fc & df2$pvs < cutoff.pvs] <- "Down"
# add a column of NAs to df2 titled genelabel that will contain names of differentially expressed genes
df2$genelabel <- NA
df2$genelabel[df2$diffexpressed != 'NO'] <- rownames(df2)[df2$diffexpressed != 'NO']
plt <- ggplot(df2, aes(x=log2fc,y=-log10(pvs), col=diffexpressed, label=genelabel)) +
geom_point() +
scale_color_manual(values = c('blue', 'black', 'red'),
name = 'Expression',
breaks = c('Down', 'NO', 'Up'),
labels = c('Down', 'N.S.', 'Up')) +
ggtitle(paste('Cluster',i,'vs. Other Clusters')) +
xlab('log2 fold change') +
ylab('-log10(p value)') +
#geom_label_repel() +
geom_vline(xintercept = c(-cutoff.log2fc, cutoff.log2fc), col='black',linetype='dashed') +
geom_hline(yintercept = -log10(cutoff.pvs), col='black',linetype='dashed') +
theme_classic()
plt
})
## save all figures
for (i in seq_len(num_center)) {
name = paste0('volcano_cluster_',i,'_vs_otherclusters.png')
plt <- volcano_plt[[i]]
ggsave(name, plot = plt, width = 10, height = 10, dpi = 300)
}
# volcano plots suggest that cluster 9 and cluster 13 could both be T cells
## compare two clusters against each other
num_cluster1 = 11
num_cluster2 = 9
cluster.of.interest <- names(which(com$cluster == num_cluster1))
cluster.other <- names(which(com$cluster != num_cluster2))
## loop through my genes and test each one
out <- sapply(genes, function(g) {
a <- mat[cluster.of.interest, g]
b <- mat[cluster.other, g]
pvs <- wilcox.test(a,b,alternative='two.sided')$p.val
log2fc <- log2(mean(a)/mean(b))
c(pvs=pvs,log2fc=log2fc)
})
## volcano plot
# run Bonferroni correction determine a p value cutoff (conservative method with low false-positive rate but high false-negative rate as a trade-off)
cutoff.pvs <- 0.05/ncol(mat) # false-positive rate would be 1-(1-0.05/ncol(mat))^ncol(mat)
cutoff.log2fc <- 2
df <- data.frame(pvs=out[1,], log2fc=out[2,])
## prepare a data frame for plotting
df2 <- df
# add a column of NAs to df2 titled diffexpressed
df2$diffexpressed <- 'NO'
# if log2fc > cutoff.log2fc and pvalue < cutoff.pvs, set as "Up"
df2$diffexpressed[df2$log2fc > cutoff.log2fc & df2$pvs < cutoff.pvs] <- "Up"
# if log2fc < -cutoff.log2fc and pvalue < cutoff.pvs, set as "Down"
df2$diffexpressed[df2$log2fc < -cutoff.log2fc & df2$pvs < cutoff.pvs] <- "Down"
# add a column of NAs to df2 titled genelabel that will contain names of differentially expressed genes
df2$genelabel <- NA
df2$genelabel[df2$diffexpressed != 'NO'] <- rownames(df2)[df2$diffexpressed != 'NO']
plt <- ggplot(df2, aes(x=log2fc,y=-log10(pvs), col=diffexpressed, label=genelabel)) +
geom_point() +
scale_color_manual(values = c('blue', 'black', 'red'),
name = 'Expression',
breaks = c('Down', 'NO', 'Up'),
labels = c('Down', 'N.S.', 'Up')) +
ggtitle(paste('Cluster',num_cluster1,'vs. Cluster',num_cluster2)) +
xlab('log2 fold change') +
ylab('-log10(p value)') +
#geom_label_repel() +
geom_vline(xintercept = c(-cutoff.log2fc, cutoff.log2fc), col='black',linetype='dashed') +
geom_hline(yintercept = -log10(cutoff.pvs), col='black',linetype='dashed') +
theme_classic()
name = paste0('volcano_cluster_',num_cluster1,'_vs_cluster_',num_cluster2,'.png')
ggsave(name, plt, width = 10, height = 10, dpi = 300)
#### plot for a figure ####
## set custom theme for plots
plot_theme <- theme_classic() +
theme(
text = element_text(size = 25),
legend.key.size = unit(0.75, 'cm')
)
## plot certain cluster on tSNE and spatial
cluster.of.interest = 9
gene.of.interest = 'CD8A'
df <- data.frame(pos, pcs$x[,1:2], emb$Y, celltype = as.factor(com$cluster), gene=mat[,gene.of.interest])
head(df)
# PCA plots
p1 <- ggplot(df, aes(x = PC1, y = PC2, col = celltype)) +
geom_point(size = 0.5) +
ggtitle('PCA (All clusters)') +
labs(color = 'Cluster') +
plot_theme
p2 <- ggplot(df, aes(x = PC1, y = PC2, col = celltype == cluster.of.interest)) +
geom_point(size = 0.5) +
scale_color_manual(values = c('lightgrey','#52BAD4'),
name = paste('Cluster',cluster.of.interest),
labels = c('FALSE','TRUE')) +
ggtitle(paste0('PCA (Cluster ',cluster.of.interest,')')) +
plot_theme
p3 <- ggplot(df, aes(x = PC1, y = PC2, col = gene)) +
geom_point(size = 0.5) +
scale_color_gradient(low='lightgrey', high='#52BAD4', name = gene.of.interest) +
ggtitle(paste0('PCA (',gene.of.interest,')')) +
plot_theme
# tSNE plots
p4 <- ggplot(df, aes(x = X1, y = X2, col = celltype)) +
geom_point(size = 0.5) +
ggtitle('tSNE (All clusters)') +
labs(color = 'Cluster') +
plot_theme
p5 <- ggplot(df, aes(x = X1, y = X2, col = celltype == cluster.of.interest)) +
geom_point(size = 0.5) +
scale_color_manual(values = c('lightgrey','#52BAD4'),
name = paste('Cluster',cluster.of.interest),
labels = c('FALSE','TRUE')) +
ggtitle(paste0('tSNE (Cluster ',cluster.of.interest,')')) +
plot_theme
p6 <- ggplot(df, aes(x = X1, y = X2, col = gene)) +
geom_point(size = 0.5) +
scale_color_gradient(low='lightgrey', high='#52BAD4', name = gene.of.interest) +
ggtitle(paste0('tSNE (',gene.of.interest,')')) +
plot_theme
# spatial plots
p7 <- ggplot(df, aes(x = x_centroid, y = y_centroid, col = celltype)) +
geom_point(size = 0.5) +
ggtitle('Spatial (All clusters)') +
labs(color = 'Cluster') +
plot_theme
p8 <- ggplot(df, aes(x = x_centroid, y = y_centroid, col = celltype == cluster.of.interest)) +
geom_point(size = 0.5) +
scale_color_manual(values = c('lightgrey','#52BAD4'),
name = paste('Cluster',cluster.of.interest),
labels = c('FALSE','TRUE')) +
ggtitle(paste0('Spatial (Cluster ',cluster.of.interest,')')) +
plot_theme
p9 <- ggplot(df, aes(x = x_centroid, y = y_centroid, col = gene)) +
geom_point(size = 0.5) +
scale_color_gradient(low='lightgrey', high='#52BAD4', name = gene.of.interest) +
ggtitle(paste0('Spatial (',gene.of.interest,')')) +
plot_theme
## volcano plot
p10 <- volcano_plt[[cluster.of.interest]] +
geom_label_repel(size = 7) +
plot_theme
lay <- rbind(c(1,2,3,10,10,10),
c(4,5,6,10,10,10),
c(7,8,9,10,10,10))
result <- grid.arrange(p1, p2, p3, p4, p5, p6, p7, p8, p9, p10, layout_matrix = lay)
## save plot
ggsave("hw5_gaihara1.png", plot = result, width = 40, height = 15, dpi = 300)
#### references ####
# information on sapply(): https://www.guru99.com/r-apply-sapply-tapply.html
# return multiple output for apply(): https://stackoverflow.com/questions/12300565/return-multiple-values-from-apply-function-in-r
# volcano plots: https://biocorecrg.github.io/CRG_RIntroduction/volcano-plots.html
# linetype: http://sape.inf.usi.ch/quick-reference/ggplot2/linetype
# bonferroni correction: https://www.aaos.org/aaosnow/2012/apr/research/research7/#:~:text=The%20Bonferroni%20correction%20is%20an,number%20of%20comparisons%20being%20made.
# bonferroni correction 2: https://www.youtube.com/watch?v=HLzS5wPqWR0&ab_channel=TopTipBio
# ggrepel package (in Japanese): https://id.fnshr.info/2017/03/19/ggrepel/
r#:~:text=Example%201%3A%20Remove%20All%20Legends%20in%20ggplot2&text=We%20simply%20had%20to%20specify,get%20rid%20of%20both%20legends.
(do you think my data visualization is effective? why or why not?)