Identifying B cell markers in imaging dataset

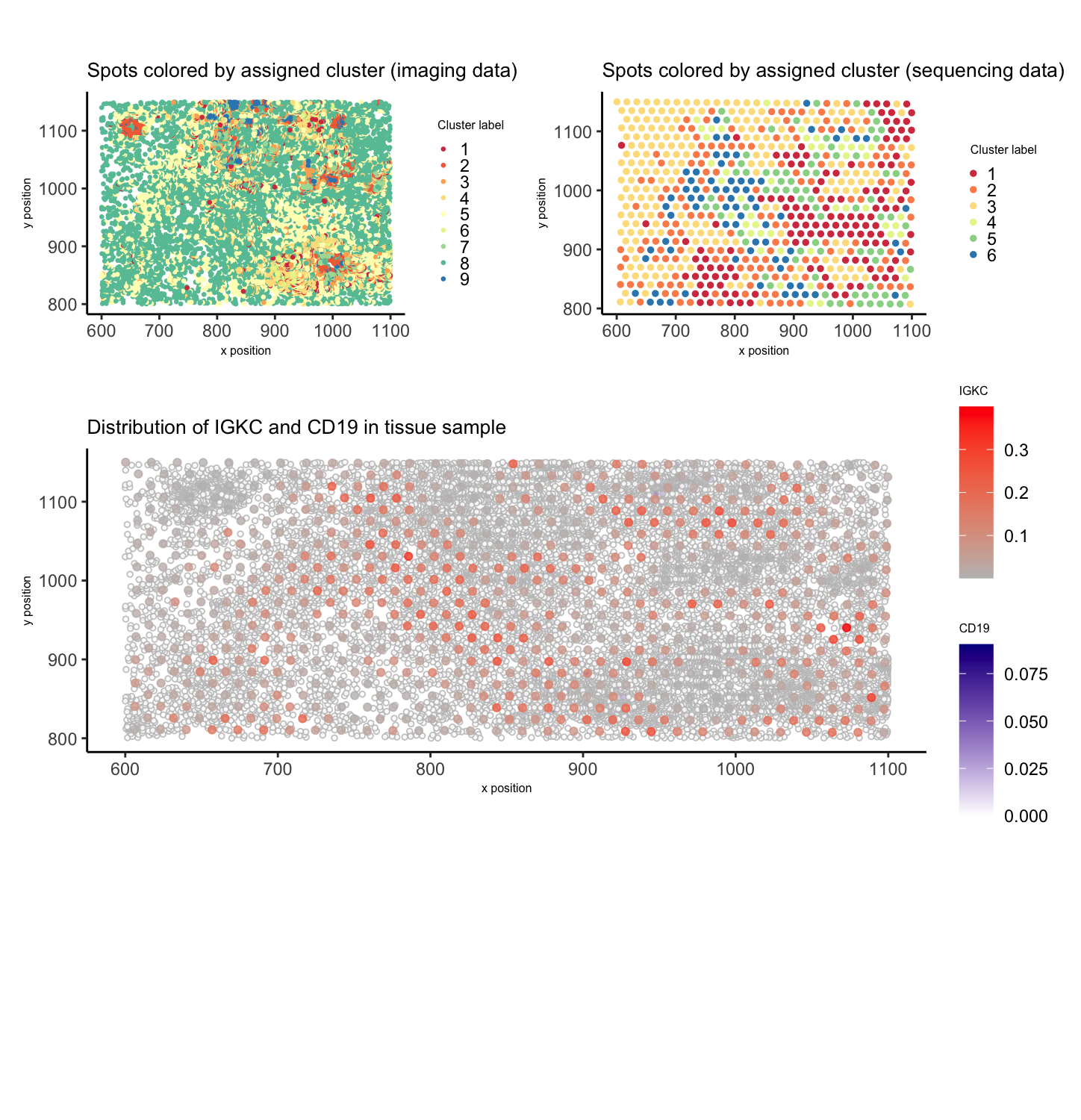
To begin analyzing the imaging dataset, I decided to normalize by cells’ areas, rather than use count-based normalization. Afterwards, I clustered my normalized gene expression data using k-means and determined 9 clusters to be optimal, based on my elbow plot. This was different from my chosen number of clusters with the sequencing data (k = 6), which reflects the increased granularity obtained with imaging data. Because there are so many more data points (cells), it makes sense that k-means clustering will identify more nuances between cells and therefore segregate the data into more clusters.
I visualized the clustering results in physical space with both the imaging and sequencing data, and I think the differences are striking. They demonstrate how the spot-based data cannot accurately capture the cell-level groupings.
This led me to question whether the B cell population I had identified in HW3 would be observed in the imaging dataset. Unfortunately, most of the upregulated genes I had looked at in my chosen cluster were not included in the imaging data. Therefore, I used another common B cell marker, CD19 (1), that was present in the imaging dataset but not in the top 1000 genes of the sequencing dataset. I wanted to visualize the overlap between IGKC in the sequencing dataset and CD19 in the imaging dataset, both associated with B cells (2).
Unfortunately, the expression of CD19 was very low throughout the tissue and therefore, trends did not line up with IGKC. There are a few possible explanations. It is possible that the B cell cluster identified with the sequencing data is present in the imaging data, but not having the same genes for analysis (ex. IGKC, IGHA1, etc.) in the imaging data means I had to use another B cell marker (CD19). However, the overall low CD19 expression makes it a poor choice for comparison with IGKC, which was highly expressed in the sequenced spots.
References:
- Wang, K., Wei, G. & Liu, D. CD19: a biomarker for B cell development, lymphoma diagnosis and therapy. Exp Hematol Oncol 1, 36 (2012). https://doi.org/10.1186/2162-3619-1-36
- ThermoFisher Scientific. Kappa Light Chain/IGKC (B-Cell Marker) Recombinant Rabbit Monoclonal Antibody (KLC2886R). https://www.thermofisher.com/antibody/product/Kappa-Light-Chain-IGKC-B-Cell-Marker-Antibody-clone-KLC2886R-Recombinant-Monoclonal/3514-RBM14-P0
Code (paste your code in between the ``` symbols)
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
library(gganimate)
library(ggplot2)
library(Rtsne)
library(patchwork)
#### setup ####
file <- '~/Desktop/GDV/pikachu.csv.gz'
file_eevee <- '~/Desktop/GDV/genomic-data-visualization-2025/data/eevee.csv.gz'
data <- read.csv(file)
data_eevee <- read.csv(file_eevee)
pos <- data[,5:6]
gexp <- data[,7:ncol(data)]
rownames(pos) <- data$cell_id
rownames(gexp) <- data$cell_id
pos_eevee <- data_eevee[,3:4]
gexp_eevee <- data_eevee[,5:ncol(data_eevee)]
rownames(pos_eevee) <- data_eevee$barcode
rownames(gexp_eevee) <- data_eevee$barcode
topgenes <- names(sort(colSums(gexp_eevee), decreasing=TRUE)[1:1000])
gexp_eevee_top <- gexp_eevee[,topgenes]
## normalization strategy: can change
norm_gexp_eevee_top <- gexp_eevee_top/rowSums(gexp_eevee_top)
#### normalize gexp values by cell area this time ####
norm_gexp = gexp / data$cell_area
topgenes <- names(sort(colSums(gexp), decreasing=TRUE))
norm_gexp_top <- norm_gexp[,topgenes] # ordering cols by most expressed genes
#### PCA scree plot ####
pcs <- prcomp(norm_gexp_top, scale. = TRUE)
# visualize scree plot: is looking at top 2 PCs reasonable?
scree_df <- data.frame(sdev = pcs$sdev, index=1:length(pcs$sdev))
scree_plt <- ggplot(scree_df, aes(x = index, y = sdev)) + geom_point()
top_PCs_scree_plt <- ggplot(scree_df[1:20,], aes(x = index, y = sdev)) +
geom_point() +
theme_classic() +
theme(plot.title = element_text(size = 10),
axis.title.x = element_text(size = 6),
axis.title.y = element_text(size = 6),
legend.title = element_text(size = 6)) +
labs(x = 'PC index', y = 'Standard deviation', title = 'Scree Plot for PCs')
#### clustering on norm_gexp_top and clusters in space ####
# kmeans clustering on normalized gexp vals
ks <- c(2,3,4,5,6,7,8,9,10,11,12)
set.seed(10)
totws <- sapply(ks, function(k) {
print(k)
clus <- kmeans(norm_gexp_top, centers = k)
return(clus$tot.withinss)
})
totws_df <- data.frame(k = ks, totw = totws)
# elbow plot
elbow_plt <- ggplot(totws_df, aes(x = k, y = totw)) +
geom_point() +
theme_classic() +
theme(plot.title = element_text(size = 10),
axis.title.x = element_text(size = 6),
axis.title.y = element_text(size = 6),
legend.title = element_text(size = 6)) +
labs(y = 'Total Withiness',
title = 'Elbow plot: determining # of clusters')
# using labels w/ 9 clusters
my_colors <- setNames(RColorBrewer::brewer.pal(9, "Spectral"),
c(1,2,3,4,5,6,7,8,9))
clus_labs <- (kmeans(norm_gexp_top, centers = 9))$cluster
# plotting spots in physical space, colored by cluster
clus_in_space <- ggplot(pos, aes(x = aligned_x, y = aligned_y,
color = as.factor(clus_labs))) +
geom_point(size = 0.5) +
theme_classic() +
theme(plot.title = element_text(size = 10),
axis.title.x = element_text(size = 6),
axis.title.y = element_text(size = 6),
legend.title = element_text(size = 6),
legend.key.size = unit(0.2,'cm')) +
scale_color_manual(values = my_colors) +
labs(x = 'x position', y = 'y position',
title = 'Spots colored by assigned cluster (imaging data)',
color = 'Cluster label')
#### plotting spots in PC space, colored by cluster ####
clus_in_PC_space <- ggplot(pcs_df, aes(x = PC1, y = PC2,
color = as.factor(clus_labs))) +
geom_point(size = 0.5) +
theme_classic() +
theme(plot.title = element_text(size = 10),
axis.title.x = element_text(size = 6),
axis.title.y = element_text(size = 6),
legend.title = element_text(size = 6),
legend.key.size = unit(0.1,'cm')) +
scale_color_manual(values = my_colors) +
geom_rect(aes(xmin = -14, xmax = 0, ymin = -5, ymax = 12),
fill = "transparent", color = "green", size = 1.5) +
labs(title = 'Spots colored by assigned cluster in PC space',
color = 'Cluster label')
#### exploring B cells in img dataset: CD19 and IGKC ####
pos_CD19 <- cbind(pos, 'Gene' = norm_gexp$CD19, 'Label' = 'Img')
pos_eevee_IGKC <- cbind(pos_eevee,
'Gene' = norm_gexp_eevee_top$IGKC, 'Label' = 'Seq')
CD19_and_IGKC <- ggplot() + geom_point(aes(x = aligned_x, y = aligned_y, fill = Gene),
data = pos_CD19, shape = 21, size = 1, color = 'grey',
alpha = 0.8) +
geom_point(aes(x = aligned_x, y = aligned_y,
color = Gene), data = pos_eevee_IGKC, alpha = 0.8) +
scale_color_gradient(low = "grey", high = "red") +
scale_fill_gradient(low = "white", high = "darkblue") +
theme_classic() +
theme(plot.title = element_text(size = 10),
axis.title.x = element_text(size = 6),
axis.title.y = element_text(size = 6),
legend.title = element_text(size = 6)) +
labs(color = 'IGKC', fill = 'CD19', x = 'x position',
y = 'y position',
title = 'Distribution of IGKC and CD19 in tissue sample')
CD19_in_space <- ggplot(pos, aes(x = aligned_x, y = aligned_y,
color = norm_gexp_top$CD19)) +
geom_point(size = 0.5) +
theme_classic() +
theme(plot.title = element_text(size = 10),
axis.title.x = element_text(size = 6),
axis.title.y = element_text(size = 6),
legend.title = element_text(size = 6),
legend.key.size = unit(0.2,'cm')) +
scale_color_gradient(low = 'grey', high = 'blue')
labs(x = 'x position', y = 'y position',
title = 'Spots colored by CD19 expression', color = 'CD19')
IGKC_in_space <- ggplot(pos_eevee, aes(x = aligned_x, y = aligned_y,
color = norm_gexp_eevee_top$IGKC)) +
geom_point(size = 1) +
theme_classic() +
theme(plot.title = element_text(size = 10),
axis.title.x = element_text(size = 6),
axis.title.y = element_text(size = 6),
legend.title = element_text(size = 6),
legend.text = element_text(size = 6)) +
scale_color_gradient(high = 'red', low = 'grey') +
labs(x = 'x position', y = 'y position',
title = 'Spots colored by IGKC expression',
color = 'IGKC')
#### bringing back eevee dataset ####
# clustering on norm_gexp_top
set.seed(10)
ks <- c(2,3,4,5,6,7,8,9,10)
totws <- sapply(ks, function(k) {
print(k)
clus <- kmeans(norm_gexp_eevee_top, centers = k)
return(clus$tot.withinss)
})
totws_df <- data.frame(k = ks, totw = totws)
# using labels w/ 6 clusters
eevee_clus_labs <- (kmeans(norm_gexp_eevee_top, centers = 6))$cluster
# plotting spots in physical space, colored by cluster
eevee_clus_in_space <- ggplot(pos_eevee, aes(x = aligned_x, y = aligned_y,
color = as.factor(eevee_clus_labs))) +
geom_point(size = 1) +
theme_classic() +
theme(plot.title = element_text(size = 10),
axis.title.x = element_text(size = 6),
axis.title.y = element_text(size = 6),
legend.title = element_text(size = 6),
legend.key.size = unit(0.1,'cm')) +
scale_color_brewer(palette="Spectral") +
labs(x = 'x position', y = 'y position',
title = 'Spots colored by assigned cluster (sequencing data)',
color = 'Cluster label')
#### plot with patchwork ####
pcs_plt <- pcs_plt + coord_fixed(ratio = 0.5)
clus_in_space <- clus_in_space + coord_fixed(ratio = 1)
elbow_plt <- elbow_plt + coord_fixed(ratio = 2)
eevee_clus_in_space <- eevee_clus_in_space + coord_fixed(ratio = 1)
panels <- ((clus_in_space + eevee_clus_in_space) / CD19_and_IGKC) +
plot_layout(widths = c(2, 2, 2), heights = c(2, 2, 2))
panels