Analyzing Immune Cell Clusters in CODEX Dataset

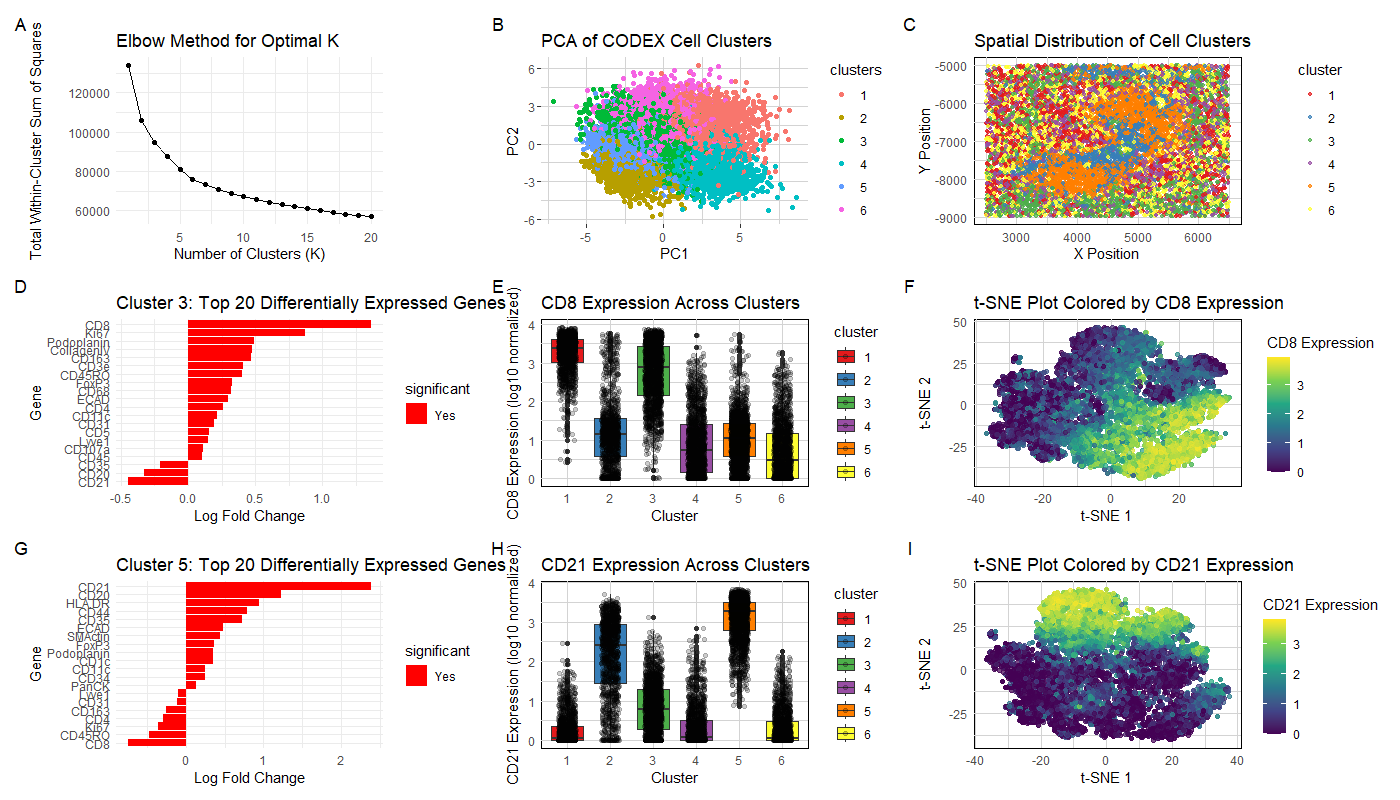
Visualization Summary In this visualization, I analyzed two cell types within the CODEX dataset: T cells and B cells. First, the genes in the dataset were normalized, log-transformed, and clustered (K = 6). The ideal K was determined through plotting the total-withiness scores for K = [1, 20]. Then, based on the elbow method heuristic, the optimal number of clusters was determined to be 6 (Figure A). To understand gene clustering and expression similarity profiles, and to reduce the dimensionality of the dataset, principal component analysis was performed (Figure B). A spatial analysis of the clusters was also visualized (Figure C). Clusters 3 and 5 were chosen for further investigation and differential gene expression analysis was performed on both clusters. A CD8 phenotype was identified in Cluster 3 (shown by Figure E, F), and a CD21 phenotype was identified in Cluster 5 (Figure H, I).
CD8 has long been classified as having a cytotoxic T cell phenotype [1], and CD21 (combined with CD20) which were both highly differentially expressed are associated with B cells [2]. Due to the discovery of these cell phenotypes, the CODEX dataset contains white pulp tissue which refers to lymphatic tissue (i.e., produces lymphocytes) [3]. Both T cells and B cells are lymphocytes.
References: [1] Koh, C. H., Lee, S., Kwak, M., Kim, B. S., & Chung, Y. (2023). CD8 T-cell subsets: heterogeneity, functions, and therapeutic potential. Experimental & molecular medicine, 55(11), 2287–2299. https://doi.org/10.1038/s12276-023-01105-x [2] Rahe, M. C., Dvorak, C. M. T., Wiseman, B., Martin, D., & Murtaugh, M. P. (2020). Establishment and characterization of a porcine B cell lymphoma cell line. Experimental cell research, 390(2), 111986. https://doi.org/10.1016/j.yexcr.2020.111986 [3] Lewis, S. M., Williams, A., & Eisenbarth, S. C. (2019). Structure and function of the immune system in the spleen. Science immunology, 4(33), eaau6085. https://doi.org/10.1126/sciimmunol.aau6085
Code (paste your code in between the ``` symbols)
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
269
270
271
272
273
274
275
276
277
278
279
280
281
282
283
284
285
286
287
288
289
290
291
292
293
294
295
296
297
298
299
300
library(ggplot2)
library(dplyr)
library(tidyr)
library(RColorBrewer)
library(patchwork)
library(Rtsne)
file <- 'codex_spleen_3.csv.gz'
data <- read.csv(file)
data[1:5,1:10]
## K Means Clustering + Principal Component Analysis
pos <- data[, 2:3]
rownames(pos) <- data$cell_id
gexp <- data[, 5:ncol(data)]
rownames(gexp) <- data$barcode
gene_norm <- log10(gexp * 10000 / rowSums(gexp) + 1)
# Compute Total Withiness
compute_wcss <- function(data, k) {
km <- kmeans(data, centers = k, nstart = 10)
return(km$tot.withinss)
}
# Compute WCSS for a range of K values
k_values <- 1:20 # You can adjust this range as needed
wcss_values <- sapply(k_values, function(k) compute_wcss(gene_norm, k))
# Create a data frame for plotting
elbow_data <- data.frame(K = k_values, WCSS = wcss_values)
# Plot the elbow curve
elbow_plot <- ggplot(elbow_data, aes(x = K, y = WCSS)) +
geom_line() +
geom_point() +
labs(title = "Elbow Method for Optimal K",
x = "Number of Clusters (K)",
y = "Total Within-Cluster Sum of Squares") +
theme_minimal()
print(elbow_plot)
# K Means Clustering
com <- kmeans(gene_norm, centers=6)
clusters <- com$cluster
clusters <- as.factor(clusters)
names(clusters) <- rownames(gene_norm)
head(clusters)
pca_result <- prcomp(gene_norm, scale. = TRUE)
summary(pca_result)
df <- data.frame(pca_result$x, clusters)
p1 <- ggplot(df, aes(x = PC1, y = PC2, col = clusters)) +
geom_point() +
labs(title = "PCA of CODEX Cell Clusters") +
theme(
panel.background = element_rect(fill = "white"),
panel.grid.major = element_line(color = "lightgray"),
panel.grid.minor = element_line(color = "lightgray")
)
print(p1)
# Combine position data with cluster information
spatial_data <- data.frame(pos, cluster = clusters)
# Create a spatial plot
p2 <- ggplot(spatial_data, aes(x = x, y = y, color = cluster)) +
geom_point(size = 1, alpha = 0.7) +
scale_color_brewer(palette = "Set1") +
labs(title = "Spatial Distribution of Cell Clusters",
x = "X Position", y = "Y Position") +
theme_minimal() +
theme(
panel.background = element_rect(fill = "white"),
panel.grid.major = element_line(color = "lightgray"),
panel.grid.minor = element_line(color = "lightgray")
)
print(p2)
# Differential Gene Expression Cluster 3
# Create a binary vector for cluster 3 vs others
cluster3_vs_others <- ifelse(clusters == 3, 1, 0)
# Function to perform t-test for each gene
perform_t_test <- function(gene) {
test_result <- t.test(gene_norm[cluster3_vs_others == 1, gene],
gene_norm[cluster3_vs_others == 0, gene])
return(c(p_value = test_result$p.value,
t_statistic = test_result$statistic))
}
# Perform t-test for all genes
t_test_results <- t(sapply(colnames(gene_norm), perform_t_test))
# Calculate log fold change
log_fc <- sapply(colnames(gene_norm), function(gene) {
mean(gene_norm[cluster3_vs_others == 1, gene]) -
mean(gene_norm[cluster3_vs_others == 0, gene])
})
# Combine results
results <- data.frame(
gene = colnames(gene_norm),
log_fc = log_fc,
p_value = t_test_results[, "p_value"],
t_statistic = t_test_results[, "t_statistic.t"]
)
# Adjust p-values for multiple testing
results$adj_p_value <- p.adjust(results$p_value, method = "BH")
# Sort by adjusted p-value
results <- results[order(results$adj_p_value), ]
# Add significance column
results$significant <- ifelse(results$adj_p_value < 0.05, "Yes", "No")
top_genes <- head(results, 20)
p4 <- ggplot(top_genes, aes(x = reorder(gene, log_fc), y = log_fc, fill = significant)) +
geom_bar(stat = "identity") +
scale_fill_manual(values = c("No" = "grey", "Yes" = "red")) +
coord_flip() +
labs(title = "Cluster 3: Top 20 Differentially Expressed Genes",
x = "Gene",
y = "Log Fold Change") +
theme_minimal()
print(p4)
## Check if CD8 is unique to cluster 3
# Create a data frame with CD8 expression and cluster information
cd8_data <- data.frame(CD8 = gene_norm[, "CD8"], cluster = clusters)
# Calculate mean CD8 expression for each cluster
cd8_mean <- cd8_data %>%
group_by(cluster) %>%
summarise(mean_expression = mean(CD8))
# Create a box plot
p_cd8 <- ggplot(cd8_data, aes(x = cluster, y = CD8, fill = cluster)) +
geom_boxplot() +
geom_jitter(width = 0.2, alpha = 0.2) +
scale_fill_brewer(palette = "Set1") +
labs(title = "CD8 Expression Across Clusters",
x = "Cluster",
y = "CD8 Expression (log10 normalized)") +
theme_minimal() +
theme(
panel.background = element_rect(fill = "white"),
panel.grid.major = element_line(color = "lightgray"),
panel.grid.minor = element_line(color = "lightgray")
)
print(p_cd8)
# Perform t-SNE
set.seed(42) # for reproducibility
tsne_result <- Rtsne(gene_norm, dims = 2, perplexity = 30, verbose = TRUE, max_iter = 1000)
# Create a data frame with t-SNE results and CD8 expression
tsne_df <- data.frame(
tSNE1 = tsne_result$Y[,1],
tSNE2 = tsne_result$Y[,2],
CD8 = gene_norm[, "CD8"]
)
# Create the t-SNE plot
p_tsne_cd8 <- ggplot(tsne_df, aes(x = tSNE1, y = tSNE2, color = CD8)) +
geom_point(alpha = 0.7) +
scale_color_viridis_c() +
labs(title = "t-SNE Plot Colored by CD8 Expression",
x = "t-SNE 1",
y = "t-SNE 2",
color = "CD8 Expression") +
theme_minimal() +
theme(
panel.background = element_rect(fill = "white"),
panel.grid.major = element_line(color = "lightgray"),
panel.grid.minor = element_line(color = "lightgray")
)
print(p_tsne_cd8)
##############################
# Differential Gene Expression Cluster 5
# Create a binary vector for cluster 5 vs others
cluster5_vs_others <- ifelse(clusters == 5, 1, 0)
# Function to perform t-test for each gene
perform_t_test <- function(gene) {
test_result <- t.test(gene_norm[cluster5_vs_others == 1, gene],
gene_norm[cluster5_vs_others == 0, gene])
return(c(p_value = test_result$p.value,
t_statistic = test_result$statistic))
}
# Perform t-test for all genes
t_test_results <- t(sapply(colnames(gene_norm), perform_t_test))
# Calculate log fold change
log_fc <- sapply(colnames(gene_norm), function(gene) {
mean(gene_norm[cluster5_vs_others == 1, gene]) -
mean(gene_norm[cluster5_vs_others == 0, gene])
})
# Combine results
results <- data.frame(
gene = colnames(gene_norm),
log_fc = log_fc,
p_value = t_test_results[, "p_value"],
t_statistic = t_test_results[, "t_statistic.t"]
)
# Adjust p-values for multiple testing
results$adj_p_value <- p.adjust(results$p_value, method = "BH")
# Sort by adjusted p-value
results <- results[order(results$adj_p_value), ]
# Add significance column
results$significant <- ifelse(results$adj_p_value < 0.05, "Yes", "No")
top_genes <- head(results, 20)
p5 <- ggplot(top_genes, aes(x = reorder(gene, log_fc), y = log_fc, fill = significant)) +
geom_bar(stat = "identity") +
scale_fill_manual(values = c("No" = "grey", "Yes" = "red")) +
coord_flip() +
labs(title = "Cluster 5: Top 20 Differentially Expressed Genes",
x = "Gene",
y = "Log Fold Change") +
theme_minimal()
print(p5)
## CD21 Analysis
cd21_data <- data.frame(CD21 = gene_norm[, "CD21"], cluster = clusters)
# Calculate mean CD8 expression for each cluster
cd21_mean <- cd21_data %>%
group_by(cluster) %>%
summarise(mean_expression = mean(CD21))
# Create a box plot
p_cd21 <- ggplot(cd21_data, aes(x = cluster, y = CD21, fill = cluster)) +
geom_boxplot() +
geom_jitter(width = 0.2, alpha = 0.2) +
scale_fill_brewer(palette = "Set1") +
labs(title = "CD21 Expression Across Clusters",
x = "Cluster",
y = "CD21 Expression (log10 normalized)") +
theme_minimal() +
theme(
panel.background = element_rect(fill = "white"),
panel.grid.major = element_line(color = "lightgray"),
panel.grid.minor = element_line(color = "lightgray")
)
print(p_cd21)
# Perform t-SNE for CD21
set.seed(42) # for reproducibility
tsne_result <- Rtsne(gene_norm, dims = 2, perplexity = 30, verbose = TRUE, max_iter = 1000)
# Create a data frame with t-SNE results and CD8 expression
tsne_df <- data.frame(
tSNE1 = tsne_result$Y[,1],
tSNE2 = tsne_result$Y[,2],
CD21 = gene_norm[, "CD21"]
)
# Create the t-SNE plot
p_tsne_cd21 <- ggplot(tsne_df, aes(x = tSNE1, y = tSNE2, color = CD21)) +
geom_point(alpha = 0.7) +
scale_color_viridis_c() +
labs(title = "t-SNE Plot Colored by CD21 Expression",
x = "t-SNE 1",
y = "t-SNE 2",
color = "CD21 Expression") +
theme_minimal() +
theme(
panel.background = element_rect(fill = "white"),
panel.grid.major = element_line(color = "lightgray"),
panel.grid.minor = element_line(color = "lightgray")
)
print(p_tsne_cd21)
## Make combined plot
combined_plot <- (elbow_plot + p1 + p2 + p4 + p_cd8 + p_tsne_cd8 + p5 + p_cd21 + p_tsne_cd21) + plot_layout(ncol = 3) +
plot_annotation(tag_levels = 'A')
print(combined_plot)
ggsave("hw5_sraghav9_v1.png", combined_plot, width = 18, height = 10, units = "in", dpi = 300)