HW1: gene expression scatterplot

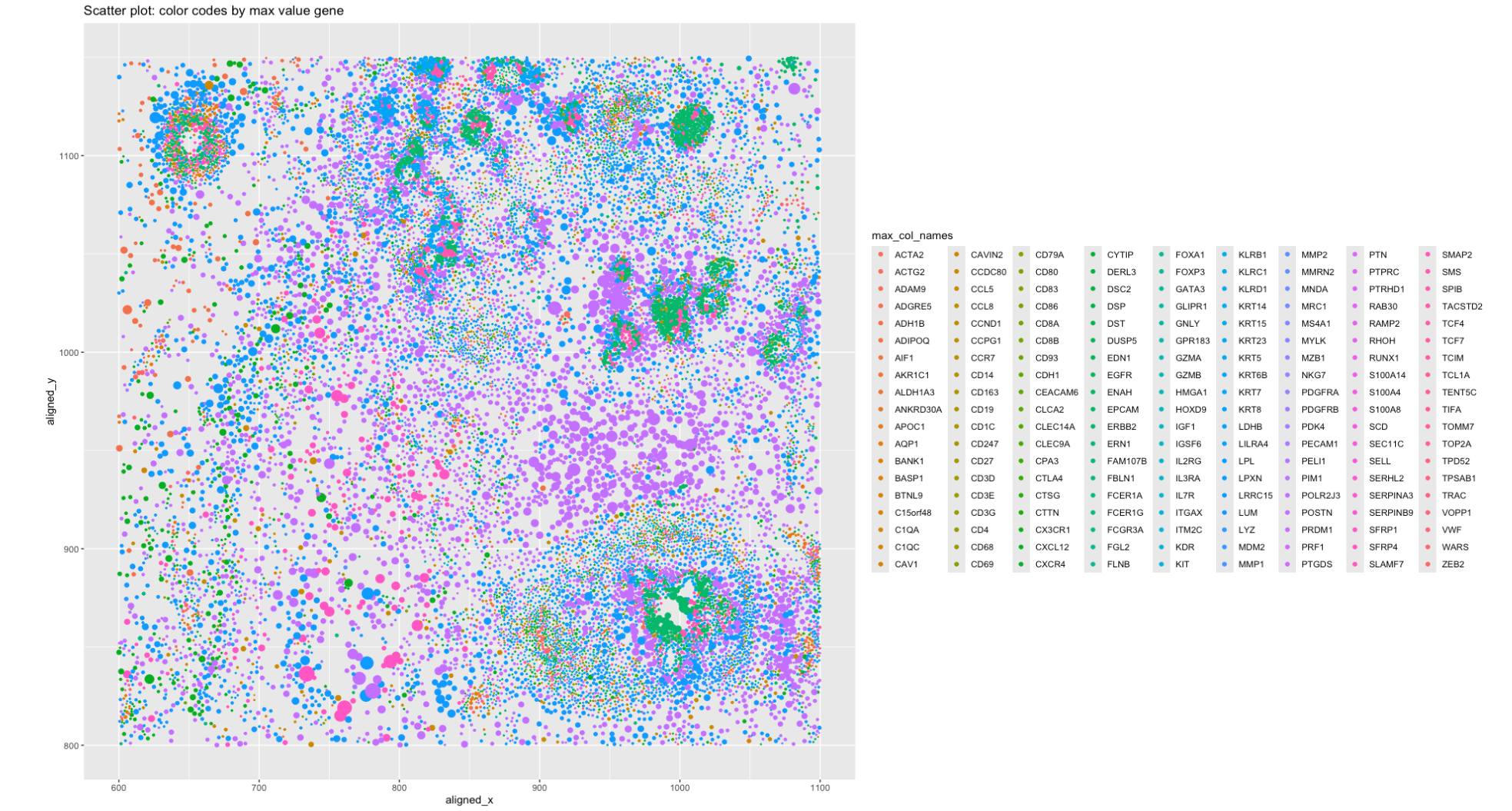
1. What data types are you visualizing?
- Spatial data of each cell, i.e the location of the cell within the section of the image, which is represented by x and y coordinates of each dot.
- Categorical data: each cell are labeled by the gene with the highest count found within the cell. (Assuming this is a characteriscs that we are interested in.) The intention is to create a 2D map to see if there are any spatial clustering can be detected.
2. What data encodings (geometric primitives and visual channels) are you using to visualize these data types?
- Used the points as geometric primitive for each cell positioned by x and y coordinates.
- Used color to encode the gene with the highest count in each cell. In retrospect, I should have limied the number of gene types to be shown or relabel them in such way that reduced the number of types. As it currently implemented, there are way too many classes that it’s hard to identify which color refers to which gene.
- Used size to suggest the counts of the gene assuming larger count means higher expression.
3. What about the data are you trying to make salient through this data visualization?
The plot is intended to show the spatial distribution of the gene with the highest count in each cell. By observing the spacial relationship, we can see cells with the same most-found gene forms clusters.
The size of the dot is used to show the “expressiveness” of the gene in each cell assuming that the gene with the highest count is the most active or saliect gene in that cell.
4. What Gestalt principles or knowledge about perceptiveness of visual encodings are you using to accomplish this?
- Similarity: The same gene is represented by the same color across the cells.
- Proximity of the dots to each other shows the spatial distribution of the gene across the cells.
5. Code (paste your code in between the ``` symbols)
1
2
3
4
5
6
7
8
9
10
11
12
13
file <- '~/Desktop/genomic-data-visualization-2025/data/pikachu.csv.gz'
data <- read.csv(file, row.names=1)
library(ggplot2)
df <- data[,7:ncol(data)]
# Find column names of max values for each row
max_col_names <- colnames(df)[apply(df, 1, which.max)]
# Find the max value for each row
max_values <- apply(df, 1, max)
# plot the data
ggplot(data) + geom_point(aes(x=aligned_x, y=aligned_y, color=max_col_names), size=max_values/25) + theme(aspect.ratio=1.0) + ggtitle("Scatter plot: color codes by max value gene")