Hw1 Data Visualization

1. What data types are you visualizing?
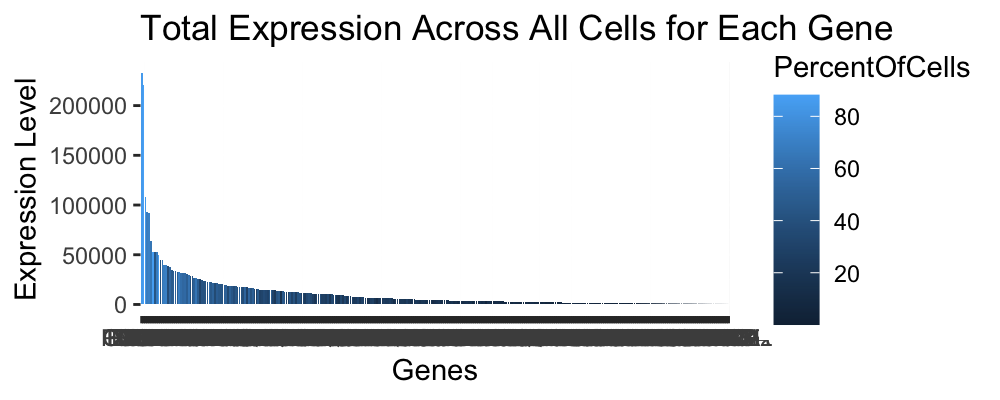
I am visualizing quantitative data of the total expression level of every gene in the panel chosen for imaging data for each cell. I am also visualizing quantitative data of the percent of cells that express each gene.
2. What data encodings (geometric primitives and visual channels) are you using to visualize these data types?
I am using the geometric primitive of bars to represent total expression of a gene. To encode the percent of cells that express each gene, I am using the visual channel of hue going from a dark blue that represents lower percent of cells that express a particular gene to a dark blue.
3. What about the data are you trying to make salient through this data visualization?
My data visualization seeks to make more salient the relationship between gene expression for each gene and the percent of cells that express that gene. The relationship shown in the graph is pretty intuitive, as it would be expected that genes that are expressed a lot are expressed by a high percentage of the cells and vice versa.
4. What Gestalt principles or knowledge about perceptiveness of visual encodings are you using to accomplish this?
I am using the continuity Gestalt principle. Sine I ordered my graphs to display in descending order for gene expression, the viewer is able to easily follow the pattern and understand the trends in the data.
5. Code (paste your code in between the ``` symbols)
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
## pikachu - imaging
file <- '/Users/anishkabhartiya/Desktop/genomic-data-visualization-2025/data/pikachu.csv.gz'
data <- read.csv(file)
## summing a column gives the total expression of that gene across all cells
head(sort(colSums(data[,7:ncol(data)]), decreasing = TRUE), n=20)
library(ggplot2)
## calculate column sums for each gene (genes start from the 7th column)
total_expression <- colSums(data[, 7:ncol(data)])
## calculate the percent of cells that express a gene
percent_expressed <- colMeans(data[, 7:ncol(data)] > 0) * 100
## create a dataframe that holds all the information with its respective gene
df <- data.frame(Gene = names(total_expression), ExpressionL = total_expression, PercentOfCells = percent_expressed)
head(df, 20)
## create bar plot
ggplot(df, aes(x = reorder(Gene, -ExpressionL),
y = ExpressionL,
fill = PercentOfCells),) +
geom_bar(stat = "identity") +
labs(
title = "Total Expression Across All Cells for Each Gene",
x = "Genes",
y = "Expression Level"
)