Visualizing tumor cells by highlighting the expression of and relationship between the ERRB2 and CCND1 genes

1. What data types are you visualizing?
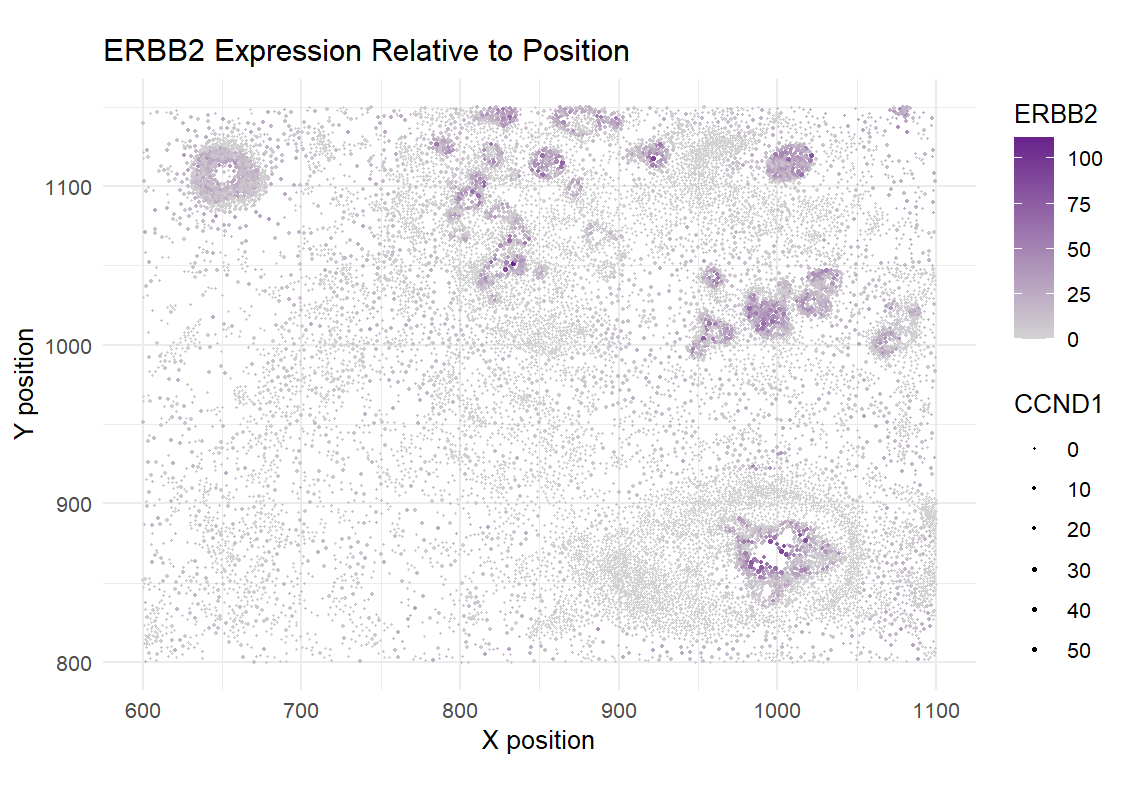
I am visualizing quantitative data of the expression counts of the ERBB2 and CCND1 genes for each cell, and spatial data of the position of cells on the tissue.
2. What data encodings (geometric primitives and visual channels) are you using to visualize these data types?
I am using the geometric primitive of points to represent each cell. It could also be argued that I used the geometric primitive of area to show expression of the CCND1 gene.
The visual channels which were used include size (a larger point size indicate higher levels of CCND1 expression), color (specifically, saturation, since a more purple saturation indicates higher ERBB2 expression, and an unsaturated lightgray indicates lower ERBB2 expression), and position (the spatial x position is encoded in the position along the x axis, and the spatial y position is encoded in the position along the y axis).
3. What about the data are you trying to make salient through this data visualization?
My data visualization hopes to highlight the relationship between the ERBB2 and CCND1 genes. They are often positively correlated, since both are involved in cancer (higher expression indicates cancerous cells). Through making this relationship salient, the regions of tumor tissue also become clear to the viewer.
4. What Gestalt principles or knowledge about perceptiveness of visual encodings are you using to accomplish this?
I am using the Gestalt principle of similarity (through color and size) to demonstrate which cells should be viewed as “grouped” together. In this case, this would mean that the cells which are more purple and larger in size, should be seen as cancerous/tumor cells. Although it could be argued that I am also using proximity, that only works in conjunction with similarly, since there are some cases of proximity and higher cell density without necessarily having high gene expression.
5. Code
file <- '~/Desktop/genomic-data-visualization-2025/data/pikachu.csv.gz'
data <- read.csv(file)
head(sort(colSums(data), decreasing = TRUE), n=20) ## finding the most expressed genes
library(ggplot2)
data %>%
ggplot() +
geom_point(aes(aligned_x, aligned_y, color=ERBB2, size = CCND1)) +
scale_size_continuous(range = c(0.2, 0.5)) +
scale_color_gradient(low="lightgray", high="darkorchid4") +
coord_equal() +
theme_minimal() +
labs(x = "X position", y = "Y position", title='ERBB2 and CCND1 Expression Relative to Position')