Expression Level Amongst Top 3 Genes

1. What data types are you visualizing?
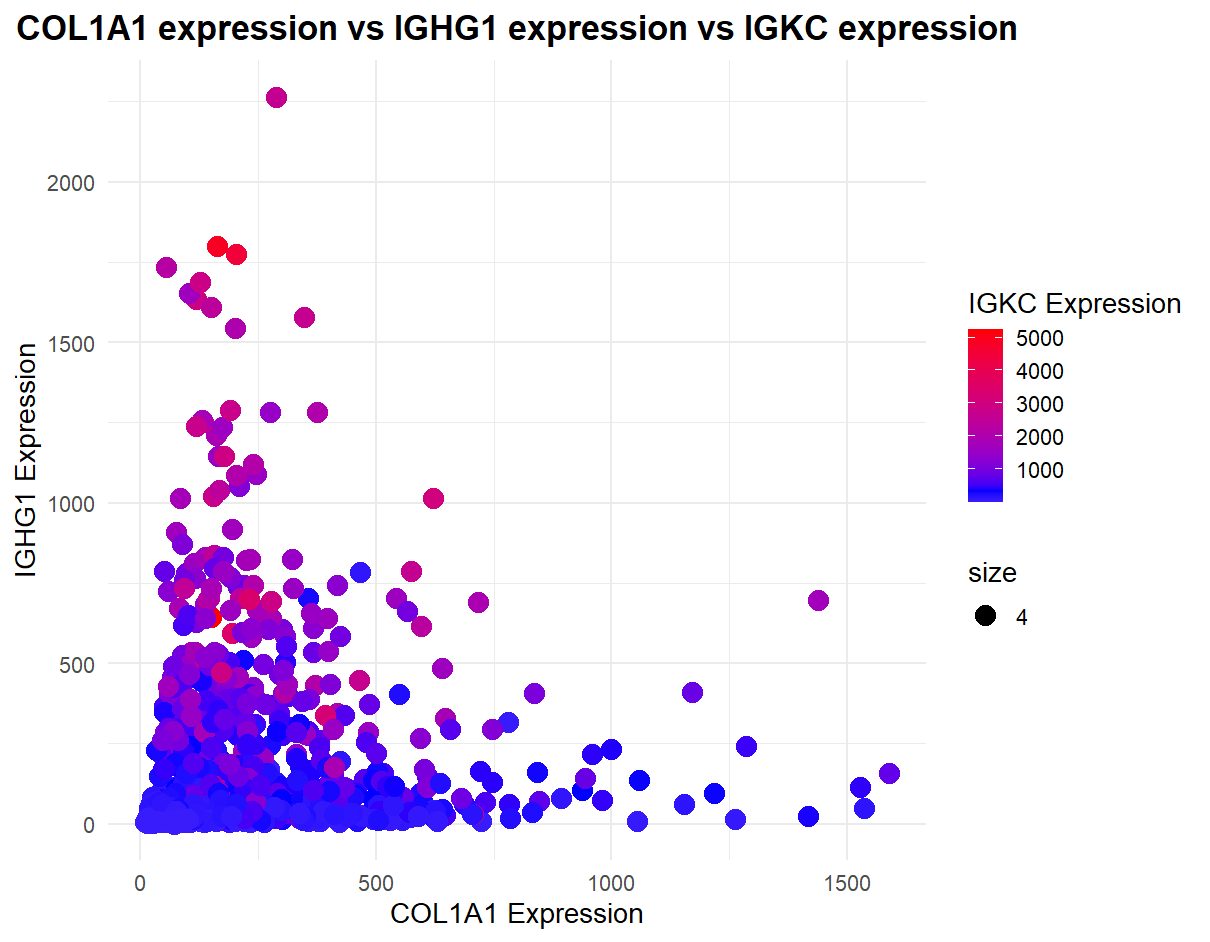
I am visualizing the top 3 genes (COL1A1, IGHG1, and IGKC) expression levels. These are all categorical data types. As in they represent qualitative characteristics that can take on numerical values, but those numbers don’t have mathematical meaning.
2. What data encodings (geometric primitives and visual channels) are you using to visualize these data types?
To visualize these data encodings, I am using geometric primatives such as points to represent each gene’s expression level. I am using color and position to represent the expression levels of each of the three genes. Position for COL1A1 and IGHG1 expression (represented on the axis) and color for IGKC expression
3. What about the data are you trying to make salient through this data visualization?
In this data, I am trying to express the relationship between the three gene’s expressions. Most importantly, I’m trying to draw the viewer to the fact that IGKC expression is highest when COL1A1 expression is low (specifically in the 0 - 500 range), indicating some sort of relationship between the two. IGHG1 and COL1A1 do not seem to have an strong expression correlation such as that. As this is categorical data, I used both position and hue (best encodings for categorical data).
4. What Gestalt principles or knowledge about perceptiveness of visual encodings are you using to accomplish this?
I am using the gestalt principle of similarity, specifically similarity in color to convey the different expression levels of IGKC. Red means high expression level whereas blue is low.
5. Code (paste your code in between the ``` symbols)
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
## eevee data
install.packages("pheatmap") # If not installed
install.packages("reshape2") # If not installed
library(pheatmap)
library(reshape2)
file <-'/Users/ishit/GenomicVis/genomic-data-visualization-2025/data/eevee.csv.gz'
data <- read.csv(file)
## data information
dim(data)
ncol(data) #how many genes in dataset
nrow(data) #how many cells
rownames(data)
colnames(data)
# picking highly expressed genes
data_numeric <- data[, sapply(data, is.numeric)]
data_filtered <- data_numeric[!rownames(data_numeric) %in% c("X", "aligned_y", "aligned_x"), ]
top_genes <- head(sort(colSums(data_filtered), decreasing = TRUE), n = 20)
print(top_genes)
# plot
library(ggplot2)
##class example
#ggplot(data) + geom_point(aes(x = IGKC, y = COL1A1, color = IGHG1)) + scale_color_gradient(low = 'lightgrey', high = 'red') + theme_bw()
## my own data visualization
ggplot(data) +
geom_point(aes(x = COL1A1, y = IGHG1, col = IGKC, size = 4)) +
scale_color_gradient2(low = "grey", mid = "blue", high = "red", midpoint = median(data$IGKC, na.rm = TRUE)) +
theme_minimal(base_size = 14) +
theme(
plot.title = element_text(hjust = 0.5, face = "bold"),
legend.position = "right"
) +
labs(title = "COL1A1 expression vs IGHG1 expression vs IGKC expression",
x = "COL1A1 Expression",
y = "IGHG1 Expression",
color = "IGKC Expression")
#refrenced: https://r-statistics.co/Top50-Ggplot2-Visualizations-MasterList-R-Code.html