Correlation Between COL1A1 and COL1A2 Gene Expression Levels in the Eevee Dataset

1. What data types are you visualizing?
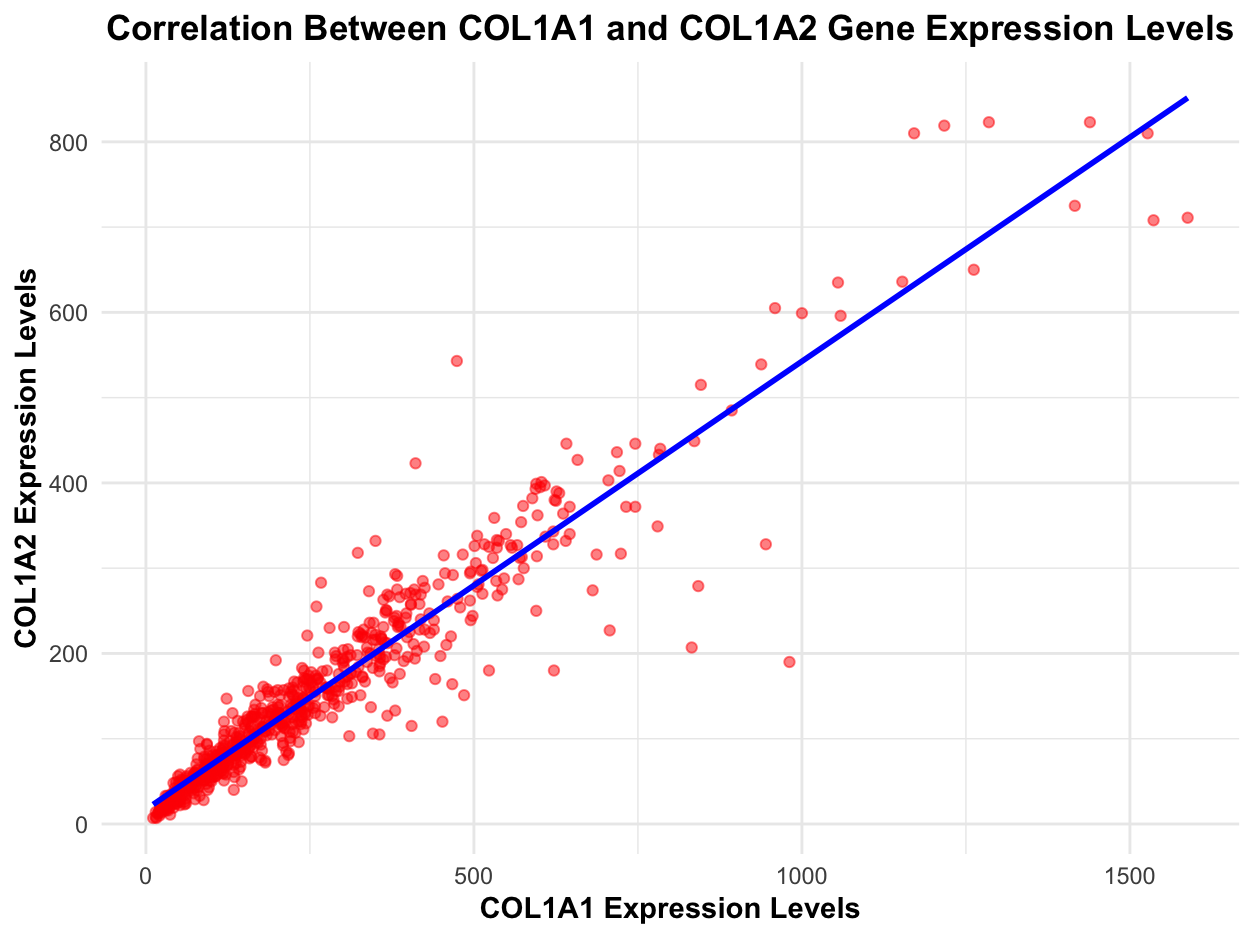
I am visualizing quantitative data of the expression levels of the COL1A1 and COL1A2 genes for each cell in the dataset.
2. What data encodings (geometric primitives and visual channels) are you using to visualize these data types?
I am using the geometric primitive of points to represent each cell. I also use the geometric primitive of a line to represent the linear regression fit and the highlight the relationship between COL1A1 and COL1A2 expression levels. To encode expression count of the COL1A1 gene, I am using the visual channel of position along the x axis. To encode expression count of the COL121 gene, I am using the visual channel of position along the y axis. To encode the type of correlation (i.e., positive or negative) and the degree of correlation (i.e., strong or weak). I also use the visual channel of color to distinguish between the points representing expression levels (red) and the trendline (blue). I also use saturation within the color channel to reduce overplotting in dense regions, making it easier to distinguish overlapping points. Darker saturation also indicates areas where points are more concentrated, highlighting the most common expression levels for both genes.
3. What about the data are you trying to make salient through this data visualization?
My data visualization seeks to make more salient the relationship between COL1A1 expression levels and COL1A2 expression levels. From the visualization, it’s clear that there is a strong positive correlation between the two genes, suggesting they are biologically interconnected in some way (although further analysis would be required to definitively make this claim). Additionally, I am try to make salient that a majority of cells follow the expected relationship between COL1A1 and COL1A2 expression levels, but there are some cells (that are far away from the trendline) that deviate from the expected relationship which may warrant more investigation into why those cells deviate.
4. What Gestalt principles or knowledge about perceptiveness of visual encodings are you using to accomplish this?
Similarity is used by coloring all points red, reinforcing that they represent the same type of data (cells), while the trendline is blue, making it visually distinct as a summary of the relationship. Proximity helps reveal natural groupings of cells with similar expression levels, where denser clusters suggest common expression patterns. Proximity to the trendline also helps identify cells that deviate from the expected relationship between COL1A1 and COL1A2 expression levels. This could be helpful because outlier cells that fall far from the trendline may represent biologically significant variations, making them candidates for further downstream analyses. Finally, continuity is applied through the regression line, guiding the viewer smoothly across the data rather than the data appearing as a random scatter.
5. Code (paste your code in between the ``` symbols)
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
file <- 'eevee.csv.gz'
data <- read.csv(file)
data[1:5, 1:10]
# Number of genes (excluding metadata columns)
num_genes <- ncol(data) - 4
# Number of cells
num_cells <- nrow(data)
# Print the results
cat("Number of genes:", num_genes, "\n")
cat("Number of cells:", num_cells, "\n")
# Sum the expression values for each gene (excluding metadata columns)
gene_expression <- colSums(data[, -(1:4)])
# Select the top 10 most highly expressed genes
top_genes <- sort(gene_expression, decreasing = TRUE)[1:10]
library(ggplot2)
# Plot Top 10 most highly expressed genes
ggplot(mapping = aes(x = reorder(names(top_genes), -top_genes), y = top_genes)) +
geom_bar(stat = "identity", fill = "skyblue") +
theme_minimal() +
labs(
title = "Top 10 Most Highly Expressed Genes",
x = "Genes",
y = "Total Expression"
) +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
# Plot Correlation Between Two Genes
ggplot(data, aes(x = COL1A1, y = COL1A2)) +
geom_point(alpha = 0.5, color = "red") +
theme_minimal() +
labs(
title = "Correlation Between COL1A1 and COL1A2 Gene Expression Levels",
x = "COL1A1 Expression Levels",
y = "COL1A2 Expression Levels"
) +
geom_smooth(method = "lm", se = FALSE, color = "blue") +
theme(
plot.title = element_text(hjust = 0.5, face = "bold"), # Center title & make bold
axis.title.x = element_text(face = "bold"), # Make x-axis label bold
axis.title.y = element_text(face = "bold") # Make y-axis label bold
)