Mean Gene Expression of Top Genes in COL Gene Group

1. What data types are you visualizing?
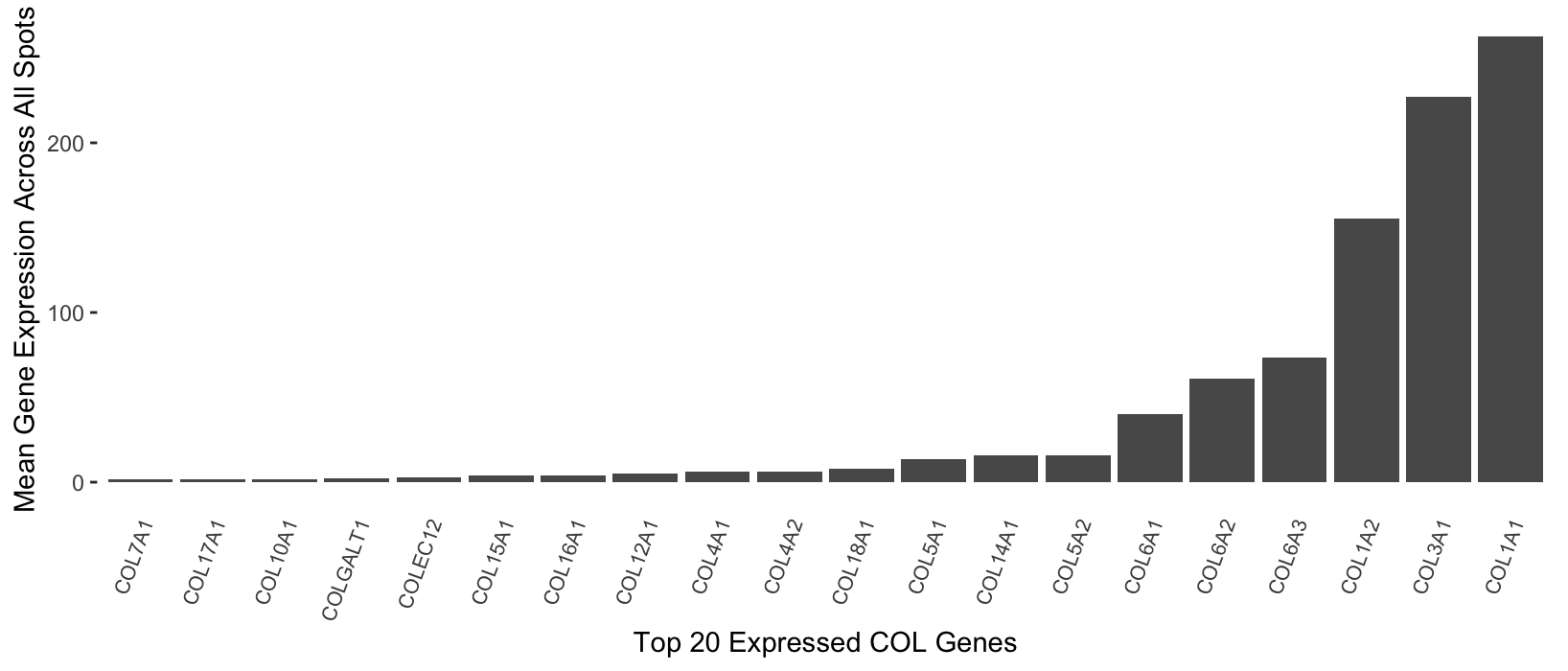
I wanted to visualize the expression of genes related to collagen in the sequencing dataset. More specifically, I looked at the mean expression of all genes starting with ‘COL’ (i.e. belonging to the collagens gene group [1]) across all spots in the sequencing dataset, where each spot coincides with a region of the human breast cancer tissue sample that was sequenced.
Therefore, I am visualizing both categorical data (different genes) and quantitative data (the mean gene expression values).
2. What data encodings (geometric primitives and visual channels) are you using to visualize these data types?
I am using the geometric primitive of lines to visualize the quantitative data, since taller lines/bars correspond to larger mean gene expression values, as well as the visual channel of position (specifically on the y-axis). I am using position on the x-axis to represent my categorical data, since the x-axis position indicates the specific gene of interest.
3. What about the data are you trying to make salient through this data visualization?
I first wanted to display a subset of the categorical data (gene names), rather than all 18085 categories/genes. I specifically tried to highlight genes related to collagen; this is because collagen is an important part of the extracellular matrix (ECM), and breast tumorigenesis and tumor cell fate are largely dependent on cell-ECM interactions [2]. I also wanted to make the mean gene expression across the entire tissue sample salient, since the individual gene expression counts per spot make it difficult to identify which collagen-associated genes are overall most highly expressed. Finally, I chose to only highlight the top 20 expressed COL genes, as opposed to all of them, in order to make these categories of genes especially salient. This approach made sense to me, as researchers may be interested in pinpointing the specific collagen subtypes that are highly expressed.
4. What Gestalt principles or knowledge about perceptiveness of visual encodings are you using to accomplish this?
Bars that have similar heights will be perceived as being in related groups (those corresponding to similarly-expressed genes), which relates to the Gestalt principle of similarity. Placing the bars in ascending order on the x-axis also allowed me to utilize the Gestalt principle of proximity, since the bars near each other are more likely to be perceived as being in similar groups (again, those with similar mean gene expression levels).
Finally, I chose to encode my quantitative data with both position and length, as well as my categorical data with position. These are some of the best ways to encode these data types for enhancing saliency and ease of perception.
References
- https://www.genenames.org/data/genegroup/#!/group/490
- https://pmc.ncbi.nlm.nih.gov/articles/PMC8911242/
Sources for coding
- https://www.datacamp.com/tutorial/sorting-in-r
- https://www.rdocumentation.org/packages/base/versions/3.6.2/topics/grep
- https://ggplot2.tidyverse.org/reference/theme.html
5. Code (paste your code in between the ``` symbols)
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
library(ggplot2)
file <- '/Users/sabahatrahman/Desktop/GDV/genomic-data-visualization-2025/data/eevee.csv.gz'
data <- read.csv(file)
pos <- data[,3:4]
gexp <- data[,5:ncol(data)]
collagens <- gexp[grep('^COL',names(gexp))] # ^ allows only strings with 'COL' at start
means <- colMeans(collagens)
means_df <- data.frame(names = names(means), means = means) # df with gene names and mean expression
means_df <- means_df[order(means_df$means, decreasing = TRUE),] # order based on means
ggplot(means_df[1:20,], aes(x = reorder(names,means), y = means)) + # only plotting top 20
geom_col() +
theme(
panel.background = element_rect(fill = 'white'), # remove gray background
axis.text.x = element_text(angle = 70, size = 8, hjust = 1), # adjust x-axis labels
axis.ticks.x=element_blank()) + # remove x-axis tick marks
labs(x = 'Top 20 Expressed COL Genes', y = 'Mean Gene Expression Across All Spots') # set axis labels