Identifying Red and White Pulp in the Spleen using CODEX Data

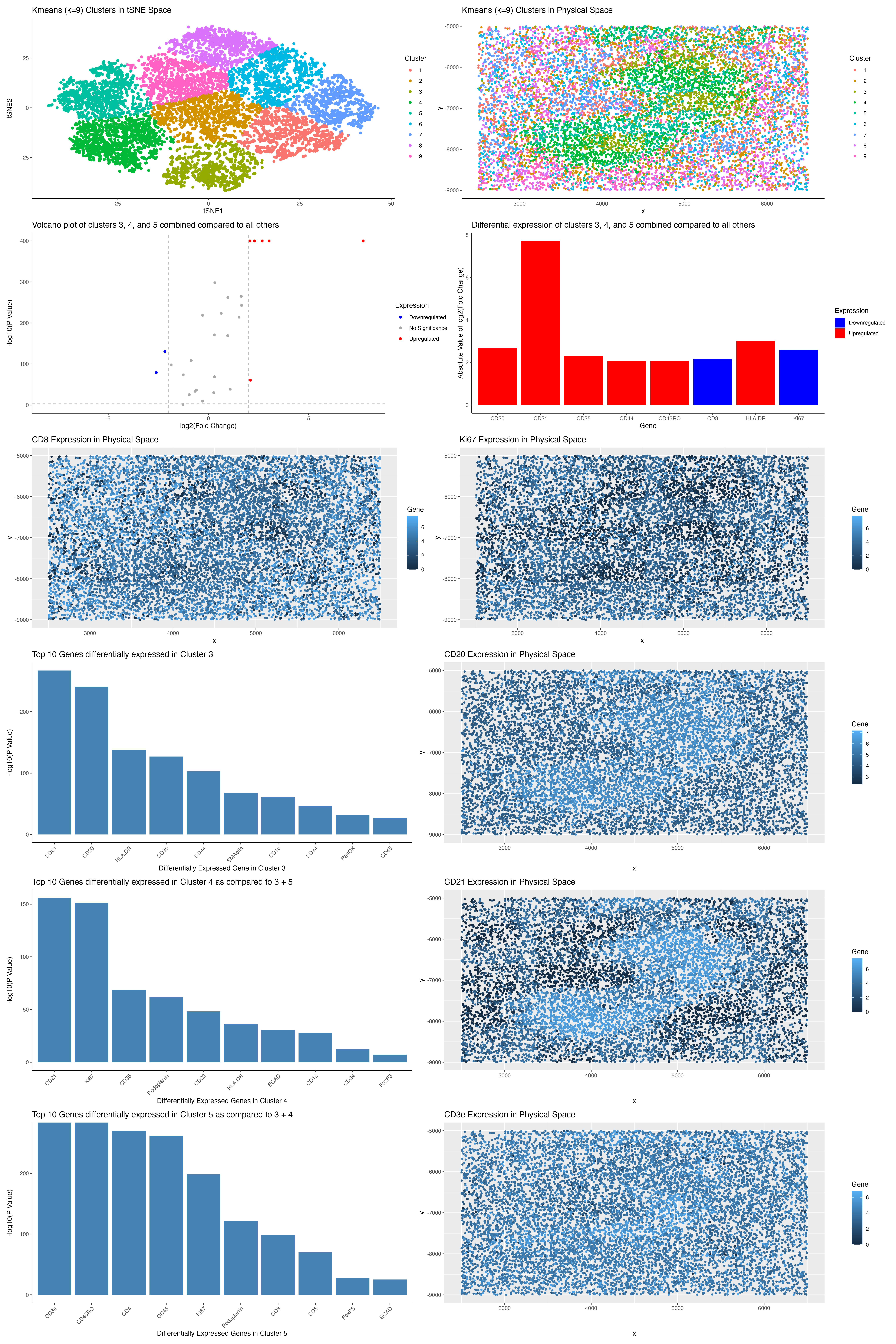
The tissue that is represented in the CODEX data is white pulp (clusters 3, 4, and 5 in my visualization) surrounded by red pulp (all other clusters). I was able to discover these tissue types by first clustering the reduced dimensionality tSNE data into 9 clusters using kmeans clustering, since 9 was found to be at the elbow of the total withiness curve. Then, I noticed when I visualized these clusters in physical space that clusters 3, 4, and 5 were localized near each other and had very few cells spread throughout the tissue slice, whereas all other clusters were composed of cells spread throughout the tissues except in the areas of cluster 3, 4, and 5 cells. Given that white pulp is much more organized (in circular regions as observed in the visualization as well) and uniform in cell types than red pulp, this observation indicated to me that clusters 3, 4, and 5 may be white pulp and the others may be red pulp (1). Thus, I stratified all the cells into two groups, those in clusters 3, 4, and 5, and those in any other cluster and performed differential protein expression on these two groups to try and identify any differential expression of marker proteins of red vs. white pulp. In doing so, I found that CD20, CD21, CD35, CD44, CD45RO, and HLA.DR are all upregulated in clusters 3, 4, and 5, and all 6 of these proteins are markers of white pulp cell types including T cells and B cells (1, 2). On the other hand, CD8 and Ki67 were found to be downregulated in clusters 3, 4, and 5, and both of these proteins are markers of red pulp cell types since CD8 is known to be highly expressed in the littoral cells in the red pulp of healthy spleens and Ki67 is a marker cell cycle activity/division that is most commonly associated with plasma cells in the red pulp in the spleen (1, 3, 4). Thus, I determined that clusters 3, 4, and 5 are white pulp, and all other clusters are red pulp.
I then aimed to identify specific cell types from the white pulp clusters. I first performed differential protein expression on cluster 3 since it appears to be the most tightly located cluster in physical space, and I identified that CD20, CD21, HLA.DR, and CD35, all of which are upregulated in B cells (3). Thus, I identified cluster 3 as B cells. Then, I decided to compare cluster 4 differential protein expression as compared to clusters 3 and 5 together to find what proteins are particularly upregulated in this cell cluster. I found that cluster 4 had even higher upregulation of CD21 and also had upregulation of Ki67, so it is likely a population of B cells that is undergoing transition from immature B cells into plasma cells before leaving the white pulp (3, 4). Finally, I also compared cluster 5 differential protein expression to clusters 3 and 4 together and found that cluster 5 had upregulation of CD3e, CD45RO, CD4, and CD45, all of which are T cell markers, and CD45RO is particularly a marker of memory T cells (3, 5). Thus, cluster 5 is likely T cells in the PALS of the white pulp.
- https://meridian.allenpress.com/aplm/article/143/9/1093/421104/Practical-Applications-in-Immunohistochemistry-An
- https://pmc.ncbi.nlm.nih.gov/articles/PMC6243053/#:~:text=The%20WP%20is%20constituted%20by,et%20al.%2C%202005
- https://www.proteinatlas.org/ENSG00000148773-MKI67/single+cell/spleen
- https://pmc.ncbi.nlm.nih.gov/articles/PMC5651088/#:~:text=A%20variety%20of%20pathologists%20had,establishing%20and%20maintaining%20humoral%20immunity.
- https://pmc.ncbi.nlm.nih.gov/articles/PMC3762710/
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
install.packages('patchwork')
set.seed(1)
data <- read.csv('~/Desktop/genomic-data-visualization-2025/data/codex_spleen_3.csv.gz', row.names=1)
data[1:5,1:5]
pos <- data[,1:2]
gexp <- data[, 4:ncol(data)]
area <- data[, 3]
head(pos)
head(gexp)
head(area)
norm <- (gexp/area)*100000 #cell area normalization
head(norm)
logexp <- log10(norm+1) #log on the normalized data
head(logexp)
library(Rtsne)
emb <- Rtsne(logexp)
ks = c(1, 2, 3, 4, 5, 7, 9, 11, 13, 15, 17, 19, 21, 23, 25, 27, 29, 31, 33, 35)
totw <- sapply(ks, function(k) {
print(k)
com <- kmeans(emb$Y, centers = k) # using the log transformed data
return(com$tot.withinss)
})
totw
plot(ks, totw)
com2 <- kmeans(emb$Y, centers = 9)
clusters2 <- com2$cluster
names(clusters2) <- rownames(gexp)
library(ggplot2)
df2 <- data.frame(tSNE1 = emb$Y[,1], tSNE2 = emb$Y[,2], cluster = factor(clusters2))
panel2 <- ggplot(df2, aes(x=tSNE1, y = tSNE2, col = cluster)) + geom_point() + theme_classic() +
labs(title = "Kmeans (k=9) Clusters in tSNE Space",
colour = 'Cluster') +
theme_classic()
panel2
df3 <- data.frame(x = pos$x, y = pos$y, clusters2)
panel3 <- ggplot(df3, aes(x=x, y = y, col = factor(clusters2))) + geom_point(size = 1) +
labs(title = "Kmeans (k=9) Clusters in Physical Space",
colour = 'Cluster') +
theme_classic()
panel3
cellsOfInterest <- names(clusters2)[clusters2 == 3 | clusters2 == 4 | clusters2 == 5]
otherCells <- names(clusters2)[clusters2 != 3 & clusters2 != 4 & clusters2 != 5]
fc <- sapply(1:ncol(norm), function(i){
genetest <- norm[,i]
names(genetest) <- rownames(norm)
foldchange <- mean(genetest[cellsOfInterest])/mean(genetest[otherCells])
})
names(fc) <- colnames(logexp)
fc
results <- sapply(1:ncol(logexp), function(i){
genetest <- logexp[,i]
names(genetest) <- rownames(logexp)
out <- wilcox.test(genetest[cellsOfInterest], genetest[otherCells], alternative = 'two.sided')
out$p.value
})
names(results) <- colnames(logexp)
sort(results, decreasing = FALSE)
df_volcano <- data.frame(gene = names(results), pval = results, fc)
reversed_pval <- -log10(df_volcano$pval)
df_volcano$reversed <- reversed_pval
df_volcano$reversed <- pmin(df_volcano$reversed, 400) #set the second number to be the top of the graph (for infinite values)
df_volcano$significantdiff <- "No Significance"
df_volcano$significantdiff[log2(fc) > 2 & df_volcano$pval < 0.05] <- "Upregulated"
df_volcano$significantdiff[log2(fc) < -2 & df_volcano$pval < 0.05] <- "Downregulated"
panel_volcano <- ggplot(df_volcano, aes(x = log2(fc), y = reversed, col = significantdiff)) +
geom_point() + theme_classic() + ylim(0, 400) + xlim(-8, 8) +
geom_vline(xintercept = -2, color = "grey", linetype = "dashed") +
geom_vline(xintercept = 2, color = "grey", linetype = "dashed") +
geom_hline(yintercept = -log(0.05), color = "grey", linetype = "dashed") +
scale_color_manual(values = c("Upregulated" = "red", "Downregulated" = "blue", "No Significance" = "darkgrey")) +
labs(title = "Volcano plot of clusters 3, 4, and 5 combined compared to all others",
x = 'log2(Fold Change)',
y= '-log10(P Value)',
colour = 'Expression')
panel_volcano
df_filtered <- df_volcano[df_volcano$significantdiff != "No Significance", ]
df_filtered <- df_filtered[order(df_filtered$fc, decreasing = TRUE), ]
significant_genes <- ggplot(df_filtered, aes(x = gene, y = abs(log2(fc)), fill = significantdiff)) +
geom_bar(stat = "identity") + theme_classic() +
scale_fill_manual(values = c("Upregulated" = "red", "Downregulated" = "blue")) +
labs(title = "Differential expression of clusters 3, 4, and 5 combined compared to all others",
x = 'Gene',
y = 'Absolute Value of log2(Fold Change)',
fill = 'Expression')
significant_genes
df_cd8 <- data.frame(x = pos$x, y = pos$y, tSNE1 = emb$Y[,1], tSNE2 = emb$Y[,2], clusters2, gene = logexp[,'CD8'])
panel_cd8 <- ggplot(df_cd8, aes(x=x, y = y, col = gene)) + geom_point(size = 1) +
labs(title = "CD8 Expression in Physical Space",
colour = 'Gene')
panel_cd8
df_ki67 <- data.frame(x = pos$x, y = pos$y, tSNE1 = emb$Y[,1], tSNE2 = emb$Y[,2], clusters2, gene = logexp[,'Ki67'])
panel_ki67 <- ggplot(df_ki67, aes(x=x, y = y, col = gene)) + geom_point(size = 1) +
labs(title = "Ki67 Expression in Physical Space",
colour = 'Gene')
panel_ki67
cellsOfInterest3 <- names(clusters2)[clusters2 == 3]
otherCells3 <- names(clusters2)[clusters2 == 4 | clusters2 == 5]
results3 <- sapply(1:ncol(logexp), function(i){
genetest <- logexp[,i]
names(genetest) <- rownames(logexp)
out <- t.test(genetest[cellsOfInterest3], genetest[otherCells3], alternative = 'greater')
out$p.value
})
names(results3) <- colnames(logexp)
sort(results3, decreasing = FALSE)
cellsOfInterest4 <- names(clusters2)[clusters2 == 4]
otherCells4 <- names(clusters2)[clusters2 == 3 | clusters2 == 5]
results4 <- sapply(1:ncol(logexp), function(i){
genetest <- logexp[,i]
names(genetest) <- rownames(logexp)
out <- t.test(genetest[cellsOfInterest4], genetest[otherCells4], alternative = 'greater')
out$p.value
})
names(results4) <- colnames(logexp)
sort(results4, decreasing = FALSE)
cellsOfInterest5 <- names(clusters2)[clusters2 == 5]
otherCells5 <- names(clusters2)[clusters2 == 3 | clusters2 == 4]
results5 <- sapply(1:ncol(logexp), function(i){
genetest <- logexp[,i]
names(genetest) <- rownames(logexp)
out <- t.test(genetest[cellsOfInterest5], genetest[otherCells5], alternative = 'greater')
out$p.value
})
names(results5) <- colnames(logexp)
sort(results5, decreasing = FALSE)
cellsOfInterest6 <- names(clusters2)[clusters2 == 3]
otherCells6 <- names(clusters2)[clusters2 != 3]
results6 <- sapply(1:ncol(logexp), function(i){
genetest <- logexp[,i]
names(genetest) <- rownames(logexp)
out <- t.test(genetest[cellsOfInterest6], genetest[otherCells6], alternative = 'greater')
out$p.value
})
names(results6) <- colnames(logexp)
sort(results6, decreasing = FALSE)
df_cluster3 <- data.frame(gene = names(sort(results6, decreasing = FALSE)[1:10]), pval = sort(results6, decreasing = FALSE)[1:10])
order <- names(sort(results6, decreasing = FALSE)[1:10])
df_cluster3$gene <- factor(df_cluster3$gene, levels = order)
panel_cluster3 <- ggplot(df_cluster3, aes(x = gene, y = -log10(pval))) +
geom_col(fill = 'steelblue') +
labs(title = "Top 10 Genes differentially expressed in Cluster 3",
x = 'Differentially Expressed Gene in Cluster 3',
y = '-log10(P Value)') +
theme_classic() +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
guides(fill = "none")
panel_cluster3
df_cluster4 <- data.frame(gene = names(sort(results4, decreasing = FALSE)[1:10]), pval = sort(results4, decreasing = FALSE)[1:10])
order <- names(sort(results4, decreasing = FALSE)[1:10])
df_cluster4$gene <- factor(df_cluster4$gene, levels = order)
panel_cluster4 <- ggplot(df_cluster4, aes(x = gene, y = -log10(pval))) +
geom_col(fill = 'steelblue') +
labs(title = "Top 10 Genes differentially expressed in Cluster 4 as compared to 3 + 5",
x = 'Differentially Expressed Genes in Cluster 4',
y = '-log10(P Value)') +
theme_classic() +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
guides(fill = "none")
panel_cluster4
df_cd21 <- data.frame(x = pos$x, y = pos$y, tSNE1 = emb$Y[,1], tSNE2 = emb$Y[,2], clusters2, gene = logexp[,'CD21'])
panel_cd21 <- ggplot(df_cd21, aes(x=x, y = y, col = gene)) + geom_point(size = 1) +
labs(title = "CD21 Expression in Physical Space",
colour = 'Gene')
panel_cd21
df_cluster5 <- data.frame(gene = names(sort(results5, decreasing = FALSE)[1:10]), pval = sort(results5, decreasing = FALSE)[1:10])
order <- names(sort(results5, decreasing = FALSE)[1:10])
df_cluster5$gene <- factor(df_cluster5$gene, levels = order)
panel_cluster5 <- ggplot(df_cluster5, aes(x = gene, y = -log10(pval))) +
geom_col(fill = 'steelblue') +
labs(title = "Top 10 Genes differentially expressed in Cluster 5 as compared to 3 + 4",
x = 'Differentially Expressed Genes in Cluster 5',
y = '-log10(P Value)') +
theme_classic() +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
guides(fill = "none")
panel_cluster5
df_cd3e <- data.frame(x = pos$x, y = pos$y, tSNE1 = emb$Y[,1], tSNE2 = emb$Y[,2], clusters2, gene = logexp[,'CD3e'])
panel_cd3e <- ggplot(df_cd3e, aes(x=x, y = y, col = gene)) + geom_point(size = 1) +
labs(title = "CD3e Expression in Physical Space",
colour = 'Gene')
panel_cd3e
library(patchwork)
panels <- (panel2 + panel3) / (panel_volcano + significant_genes) / (panel_cd8 + panel_ki67) / (panel_cluster3 + panel_cd20) / (panel_cluster4 + panel_cd21) / (panel_cluster5 + panel_cd3e) +
plot_layout(widths = c(1, 1), heights = c(1, 1, 1, 1, 1, 1))
panels
ggsave("HW5_agorham3.png", panels, width = 20, height = 30, dpi = 300)