Analysis of CODEX dataset

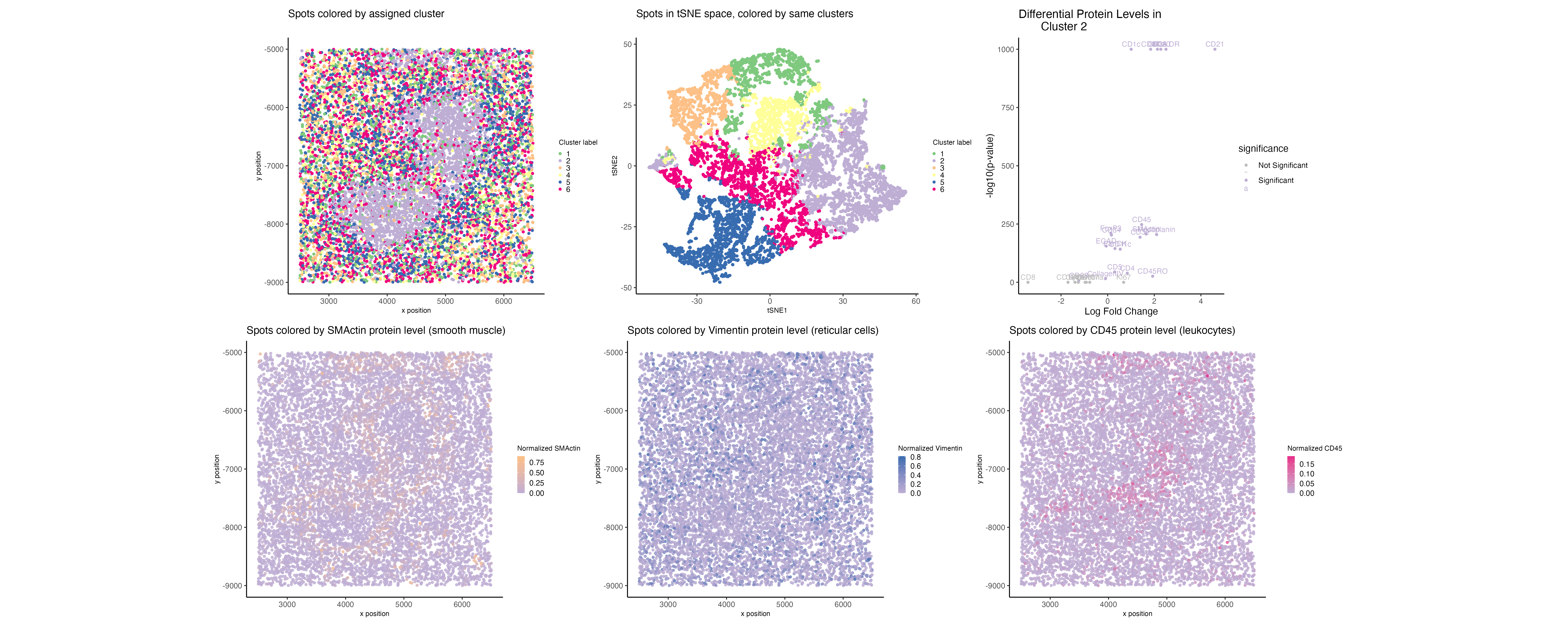
Based on the CODEX data, I hypothesize this tissue sample is taken from the white pulp region of the spleen, which is surrounded by red pulp. Some evidence/reasoning is outlined below:
- I just started by looking at the overall highest detected proteins and noticed many markers of immune cells, as well as proteins like Vimentin and SMActin (associated with endothelial/fibroblast cells and smooth muscle, respectively) [1,2].
- Based on the results of clustering via k-means on the normalized protein levels, there appears to be a clear inner region (shaped a bit like a figure-8) and a surrounding outer region. This is consistent with the anatomy of the spleen, wherein the inner region is the white pulp and the outer region is the red pulp.
- I looked at the upregulated proteins in the figure-8 region. One of these was CD45, a marker common to leukocytes and not found on RBCs [3]. This was evidence that this region could be the white pulp. Other upregulated proteins of interest included HLA-DR (expressed by cells like dendritic cells and B cells) [4] and FoxP3 (a regulatory T cell marker) [5].
- Importantly, Vimentin was not upregulated in the figure-8 region—this led me to reason that the surrounding structure could comprise many reticular cells, consistent with the red pulp region [6].
- I looked at various genes in physical space and was particularly struck by the SMActin expression pattern, which seems to line the figure-8 region. This makes sense to me, given that Pinkus et al. looked at the localization of smooth muscle in the spleen and found a circular/circumferential pattern [7].
References:
- https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/vimentin#:~:text=Vimentin%20is%20an%20intermediate%20filament%20protein%20found%20in%20many%20types,days%20to%20weeks%20of%20proliferation
- https://www.mayocliniclabs.com/test-catalog/overview/70551
- https://www.sciencedirect.com/science/article/pii/S0006497120738280#:~:text=THE%20LEUKOCYTE%20COMMON%20antigen%20(CD45)%20is%20an%20abundant%20cell%20surface,10%25%20of%20cell%20membrane%20proteins.&text=It%20is%20a%20member%20of%20the%20protein%20tyrosine%20phosphatase%20family.
- https://onlinelibrary.wiley.com/doi/10.1002/pros.20432
- https://www.nature.com/articles/nri.2017.75
- https://www.sciencedirect.com/topics/medicine-and-dentistry/spleen-cell#:~:text=Besides%20reticular%20cells%2C%20the%20red,nodules%20appended%20to%20the%20sheaths.
- https://pmc.ncbi.nlm.nih.gov/articles/PMC1888274/?page=6
Code (paste your code in between the ``` symbols)
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
library(ggplot2)
library(patchwork)
library(Rtsne)
#### read in data and normalize ####
data <- read.csv('~/Desktop/GDV/genomic-data-visualization-2025/data/codex_spleen_3.csv.gz')
pos <- data[,2:3]
areas <- data[,4]
prots <- data[,5:ncol(data)]
rownames(pos) <- data[,1]
rownames(prots) <- data[,1]
# using library size normalization #
norm_prots <- prots/rowSums(prots)
#### look for overall highest vals, formulate hypotheses ####
tot_prots <- colSums(prots)
names(tot_prots) <- colnames(prots)
tot_prots <- sort(tot_prots)
#### visualize spots and kmeans clusters ####
# kmeans
set.seed(6)
ks <- c(2,3,4,5,6,7,8,9,10,11,12)
totws <- sapply(ks, function(k) {
print(k)
clus <- kmeans(norm_prots, centers = k)
return(clus$tot.withinss)
})
totws_df <- data.frame(k = ks, totw = totws)
# elbow plot
elbow_plt <- ggplot(totws_df, aes(x = k, y = totw)) +
geom_point() +
theme_classic() +
theme(plot.title = element_text(size = 8),
axis.title.x = element_text(size = 8),
axis.title.y = element_text(size = 8),
legend.title = element_text(size = 8)) +
labs(x = 'k', y = 'Total Withiness',
title = 'Elbow plot for determining # of clusters')
print(elbow_plt)
# using labels w/ 8 clusters
clus_labs <- (kmeans(norm_prots, centers = 8))$cluster
clus_labs <- as.factor(clus_labs)
# plotting spots in physical space, colored by cluster
clus_in_space <- ggplot(pos, aes(x = x, y = y,
color = clus_labs)) +
geom_point(size = 1) +
theme_classic() +
theme(plot.title = element_text(size = 8),
axis.title.x = element_text(size = 8),
axis.title.y = element_text(size = 8),
legend.title = element_text(size = 8),
legend.key.size = unit(0.1,'cm')) +
scale_color_brewer(palette="Dark2") +
labs(x = 'x position', y = 'y position',
title = 'Spots colored by assigned cluster', color = 'Cluster label')
print(clus_in_space)
#### diff gexp analysis ####
# clusters of interest
ct6 <- names(clus_labs)[which(clus_labs == 6)]
ct6other <- names(clus_labs)[which(clus_labs != 6)]
ct5 <- names(clus_labs)[which(clus_labs == 5)]
ct5other <- names(clus_labs)[which(clus_labs != 5)]
ct1 <- names(clus_labs)[which(clus_labs == 1)]
ct1other <- names(clus_labs)[which(clus_labs != 1)]
# wilcox one-sided tests for clusters of interest
results6 <- sapply(colnames(norm_prots), function(i) {
wilcox.test(norm_prots[ct6, i], norm_prots[ct6other, i],
alternative = 'greater')$p.value ## two sided test
})
names(results6) <- colnames(norm_prots)
results6 <- sort(results6)
results5 <- sapply(colnames(norm_prots), function(i) {
wilcox.test(norm_prots[ct5, i], norm_prots[ct5other, i],
alternative = 'greater')$p.value ## two sided test
})
names(results5) <- colnames(norm_prots)
results5 <- sort(results5)
results1 <- sapply(colnames(norm_prots), function(i) {
wilcox.test(norm_prots[ct1, i], norm_prots[ct1other, i],
alternative = 'greater')$p.value ## two sided test
})
names(results1) <- colnames(norm_prots)
results1 <- sort(results1)
#### using fewer clusters ####
# using labels w/ 6 clusters
set.seed(10)
clus_labs_2 <- (kmeans(norm_prots, centers = 6))$cluster
clus_labs_2 <- as.factor(clus_labs_2)
# plotting spots in physical space, colored by cluster
clus_in_space_2 <- ggplot(pos, aes(x = x, y = y,
color = clus_labs_2)) +
geom_point(size = 1) +
theme_classic() +
theme(plot.title = element_text(size = 12),
axis.title.x = element_text(size = 8),
axis.title.y = element_text(size = 8),
legend.title = element_text(size = 8),
legend.key.size = unit(0.1,'cm')) +
scale_color_brewer(palette="Accent") +
labs(x = 'x position', y = 'y position',
title = 'Spots colored by assigned cluster', color = 'Cluster label')
print(clus_in_space_2)
#### visualize clusters in PC space ####
# plotting spots in PC space, colored by cluster
pcs <- prcomp(norm_prots)
pcs_df <- data.frame(pcs$x)
clus_in_PC_space <- ggplot(pcs_df, aes(x = PC1, y = PC2,
color = as.factor(clus_labs_2))) +
geom_point(size = 2) +
theme_classic() +
theme(plot.title = element_text(size = 8),
axis.title.x = element_text(size = 8),
axis.title.y = element_text(size = 8),
legend.title = element_text(size = 8),
legend.key.size = unit(0.1,'cm')) +
scale_color_brewer(palette="Accent") +
labs(title = 'Spots in PC space, colored by same clusters',
color = 'Cluster label')
print(clus_in_PC_space)
#### visualize clusters in tSNE space ####
emb <- Rtsne(norm_prots)
tsne_df <- data.frame(tSNE1 = emb$Y[,1], tSNE2 = emb$Y[,2])
tsne_plt <- ggplot(tsne_df, aes(x = tSNE1, y = tSNE2,
color = as.factor(clus_labs_2))) +
geom_point(size = 1) +
theme_classic() +
theme(plot.title = element_text(size = 12),
axis.title.x = element_text(size = 8),
axis.title.y = element_text(size = 8),
legend.title = element_text(size = 8),
legend.key.size = unit(0.1,'cm')) +
scale_color_brewer(palette="Accent") +
labs(title = 'Spots in tSNE space, colored by same clusters',
color = 'Cluster label')
#### diff gexp analysis w/ new clusters ####
ct1_new <- names(clus_labs_2)[which(clus_labs_2 == 2)]
ct1other_new <- names(clus_labs_2)[which(clus_labs_2 != 2)]
results1_new <- sapply(colnames(norm_prots), function(i) {
wilcox.test(norm_prots[ct1_new, i], norm_prots[ct1other_new, i],
alternative = 'greater')$p.value ## two sided test
})
names(results1_new) <- colnames(norm_prots)
results1_new <- data.frame(results1_new)
colnames(results1_new) <- 'results'
# plot volcano plt
mean_prots_ct1 <- colMeans(norm_prots[ct1_new, ])
mean_prots_other <- colMeans(norm_prots[ct1other_new, ])
logFC <- log2(mean_prots_ct1/mean_prots_other)
volcano_df <- data.frame(protein = names(logFC),
logFC = logFC,
p_val = results1_new$results)
volcano_df$pval_log <- -log10(volcano_df$p_val)
volcano_df$pval_log[is.infinite(volcano_df$pval_log)] <- 1000
volcano_df$significance <- ifelse(volcano_df$p_val < 0.01,
"Significant", "Not Significant")
volcano_plt <- ggplot(volcano_df, aes(x = logFC, y = pval_log,
color = significance,
label = protein)) +
geom_point(size = 1) +
geom_text(vjust = -0.5, size = 3) +
scale_color_manual(values = c("Significant" = "#beaed4",
"Not Significant" = "grey")) +
theme(plot.title = element_text(size = 6),
axis.title.x = element_text(size = 6),
axis.title.y = element_text(size = 6),
legend.title = element_text(size = 6)) +
theme_classic() +
labs(title = "Differential Protein Levels in
Cluster 2", x = "Log Fold Change",
y = "-log10(p-value)")
#### visualize cell types ####
CD45_cells <- ggplot(pos, aes(x = x, y = y,
color = norm_prots$CD45)) +
geom_point(size = 1, alpha = 0.8) +
theme_classic() +
theme(plot.title = element_text(size = 12),
axis.title.x = element_text(size = 8),
axis.title.y = element_text(size = 8),
legend.title = element_text(size = 8),
legend.key.size = unit(0.3,'cm')) +
scale_color_gradient(low = '#beaed4', high = '#e7298a') +
labs(x = 'x position', y = 'y position',
title = 'Spots colored by CD45 protein level (leukocytes)',
color = 'Normalized CD45')
vimentin_cells <- ggplot(pos, aes(x = x, y = y,
color = norm_prots$Vimentin)) +
geom_point(size = 1, alpha = 0.8) +
theme_classic() +
theme(plot.title = element_text(size = 12),
axis.title.x = element_text(size = 8),
axis.title.y = element_text(size = 8),
legend.title = element_text(size = 8),
legend.key.size = unit(0.3,'cm')) +
scale_color_gradient(low = '#beaed4', high = '#386cb0') +
labs(x = 'x position', y = 'y position',
title = 'Spots colored by Vimentin protein level (reticular cells)',
color = 'Normalized Vimentin')
SMA_cells <- ggplot(pos, aes(x = x, y = y,
color = norm_prots$SMActin)) +
geom_point(size = 1, alpha = 0.8) +
theme_classic() +
theme(plot.title = element_text(size = 12),
axis.title.x = element_text(size = 8),
axis.title.y = element_text(size = 8),
legend.title = element_text(size = 8),
legend.key.size = unit(0.3,'cm')) +
scale_color_gradient(low = '#beaed4', high = '#fdc086') +
labs(x = 'x position', y = 'y position',
title = 'Spots colored by SMActin protein level (smooth muscle)',
color = 'Normalized SMActin')
#### final plot ####
clus_in_space_2 <- clus_in_space_2 + coord_fixed(ratio = 1)
tsne_plt <- tsne_plt + coord_fixed(ratio = 1)
volcano_plt <- volcano_plt + coord_fixed(ratio = 0.01)
SMA_cells <- SMA_cells + coord_fixed(ratio = 1)
vimentin_cells <- vimentin_cells + coord_fixed(ratio = 1)
CD45_cells <- CD45_cells + coord_fixed(ratio = 1)
plot <- (clus_in_space_2 + tsne_plt + volcano_plt) /
(SMA_cells + vimentin_cells + CD45_cells)
ggsave("~/Desktop/hw5_srahma22.png", plot, width = 25, height = 10, dpi = 300)