EC1

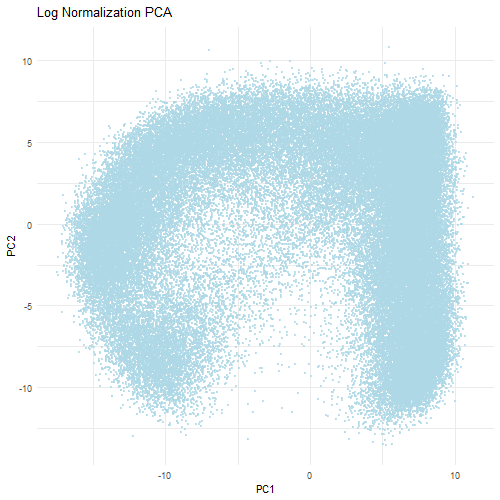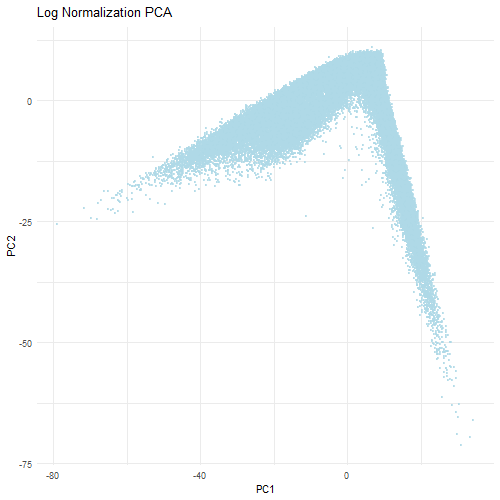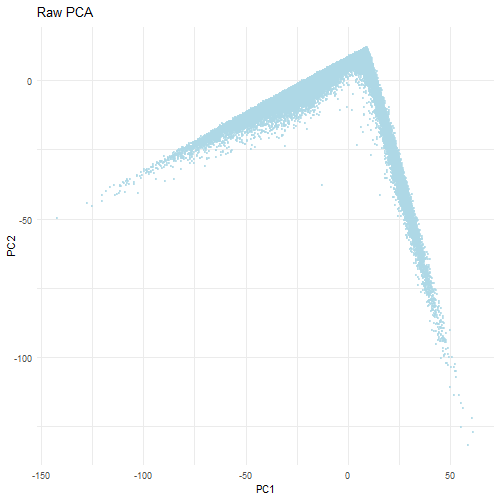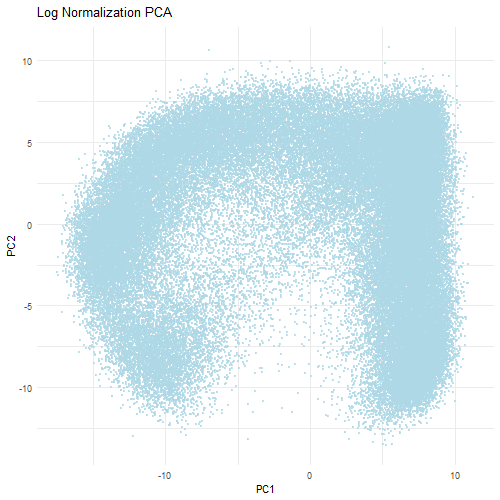
Discussion
For this homework, I wanted to answer this question: What happens if I do or not not normalize and/or transform the gene expression data (e.g. log and/or scale) prior to dimensionality reduction?
When dimensionality reduction like PCA is performed on raw gene expression counts without information, the resulting plot is usually dominated by technical variation rather than biological differences. In the raw count PCA, most of the variance is caused by differences in transcriptional counts across cells. Additionally, because PCA finds the directions of greatest variance, highly expressed genes may cause PC1 to not be reflective of the actual data. This in turn creates a skewed distriction where PC1 reflects total gene count rather than biological structure and differences. This is why the non-normalized visualization shows a long and almost triangular shaped spread along PC1 with the PC2 values being clustered near zero.
However, when the data is first normalized and log-transformed, PCA is able to find more relevant biological variation. Normalization ensures that each gene contributes fairly to the PCA analysis. The log transformation reduces the skewness of the data and stabilizes the amount of variance which prevents a small number of highly expressed genes from dominating. Therefore, the PCA plot when normalized and log-tranformed is more balanced with the data evenly distributed across the PCs. This indicates that the PCs are now reflective of the relative gene expression differences rather than technical differences that are found in the dataset. Ultimately, normalization and transformation are critical to ensure that accurate biological patterns are discerned from the data.
Code
```r
library(ggplot2) library(gganimate) library(magick) library(dplyr)
read in data
data <- read.csv(‘C:/Users/gtbud/Downloads/Xenium-IRI-ShamR_matrix.csv.gz’) rownames(data) <- data[,1] gexp <- data[, 4:ncol(data)]
raw PCA
vg_raw <- apply(gexp, 2, var) vargenes_raw <- names(sort(vg_raw, decreasing=TRUE)[1:250]) gexp_sub_raw <- gexp[, vargenes_raw]
pcs_raw <- prcomp(gexp_sub_raw, scale.=FALSE)
df_raw <- data.frame( cell = rownames(pcs_raw$x), PC1 = pcs_raw$x[,1], PC2 = pcs_raw$x[,2], state = “Raw PCA” )
normalize PCA with log
totgexp <- rowSums(gexp) mat_norm <- log10(gexp / totgexp * 1e6 + 1)
vg_norm <- apply(mat_norm, 2, var) vargenes_norm <- names(sort(vg_norm, decreasing=TRUE)[1:250]) mat_sub_norm <- mat_norm[, vargenes_norm]
pcs_norm <- prcomp(mat_sub_norm, scale.=FALSE)
df_norm <- data.frame( cell = rownames(pcs_norm$x), PC1 = pcs_norm$x[,1], PC2 = pcs_norm$x[,2], state = “Log Normalization PCA” )
df_combined <- bind_rows(df_raw, df_norm)
animate using gganimate
anim <- ggplot(df_combined, aes(x = PC1, y = PC2, group = cell)) + geom_point(color = “lightblue”, size = 0.6, alpha = 0.7) + transition_states( state, transition_length = 2, state_length = 1 ) + labs(title = ‘{closest_state}’) + ease_aes(‘cubic-in-out’) + view_follow(fixed_x = FALSE, fixed_y = FALSE) + theme_minimal()
animate(anim, nframes = 100, fps = 10, width = 400, height = 400, renderer = magick_renderer())
anim_plot <- animate( anim, nframes = 100, fps = 10, width = 500, height = 500, renderer = magick_renderer() )
anim_save( “C:/Users/gtbud/Downloads/PCA_animation.gif”, animation = anim_plot )
The beginning part of the code was done in class with Prof. Fan
AI was used to figure out download the GIF and R syntax