Identifying a kidney cortical tubule region using marker genes

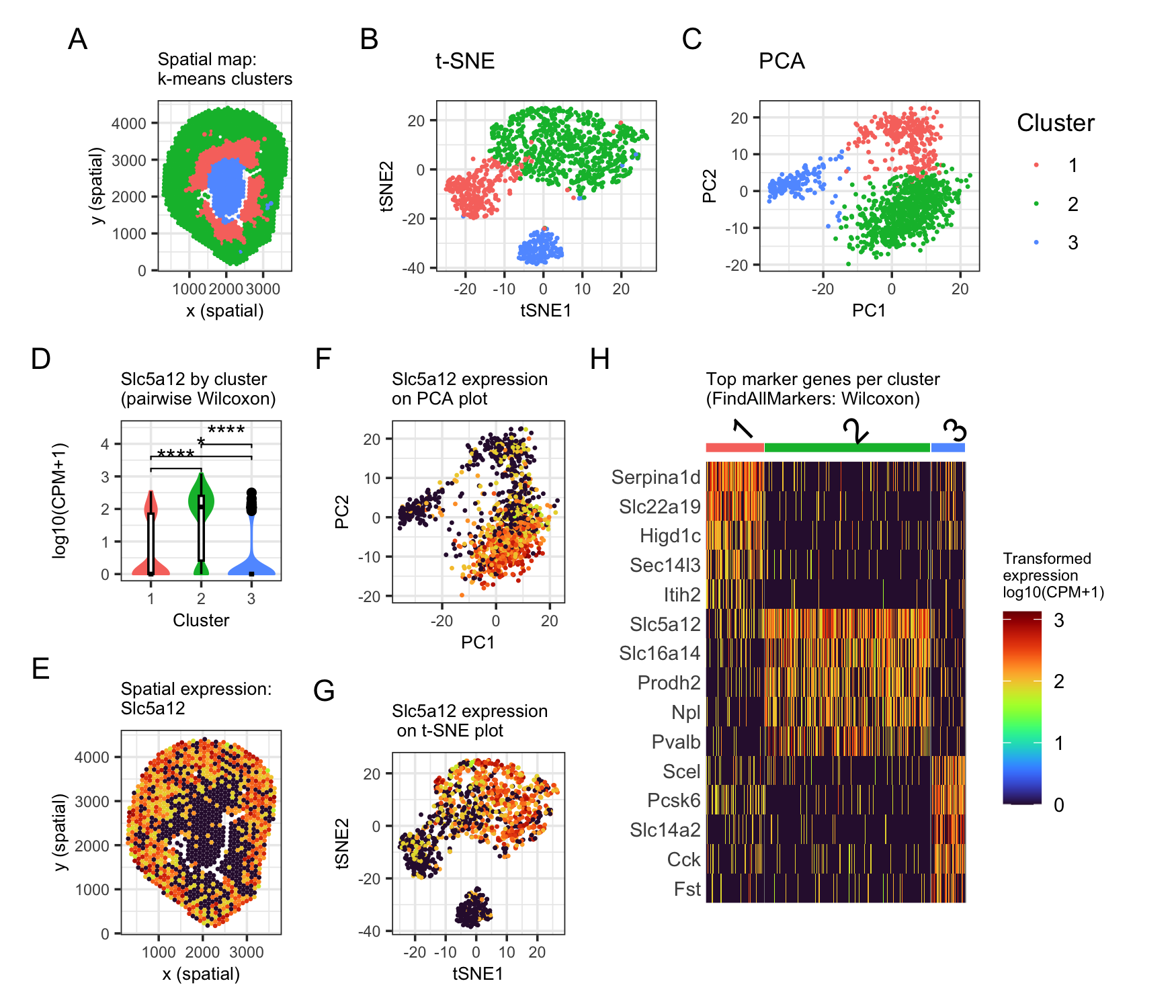
The data were normalized and log-transformed. I then ran PCA on the normalized matrix, used the scree plot of PC standard deviations to pick a safe cutoff (PC = 10) for downstream analysis, and ran t-SNE. After that, I performed k-means clustering in PC space and chose k = 3 based on the elbow plot of within-cluster variance as well as my results from previous assignments.
In the spatial plot, the clusters form a ring structure: cluster 2 (green) is on the outside, cluster 1 (red) is in the middle ring, and cluster 3 (blue) is in the center. This matches kidney anatomy that the outer region is cortex, while the center is medulla (A). In reduced dimensional space, the three clusters separate well, and clusters 1 and 2 are closer to each other than cluster 3 (B, C). I focus on cluster 2 as my distinct cluster of interest. Based on literature, Slc5a12 is expressed in the cortical tubule of kidney (Park et al., 2018). I therefore visualized Slc5a12 across clusters and in space (D, E, F, G). The violin plot with pairwise Wilcoxon tests shows Slc5a12 is significantly higher in cluster 2 compared with clusters 1 and 3 (D), and the spatial plots show the same pattern (E–G).
To further support this, I used Seurat FindAllMarkers (Wilcoxon) and plotted top marker genes across clusters (H). Cluster 2’s markers include Slc5a12 and other tubule-associated transport genes, supporting that this cluster is enriched for cortical tubules, most consistent with a proximal tubule region. I also note that Pvalb is a known marker of the early distal convoluted tubule, so there might be some mixing of cortical tubule segments as each Visium spot can contain multiple nearby cell types. Overall, the spatial location, outer cortex, plus the strong Slc5a12 enrichment supports the interpretation that cluster 2 corresponds to a cortex tubule region.
Jihwan Park et al. ,Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease.Science360,758-763(2018).DOI:10.1126/science.aar2131
I used AI to debug patchwork layout and formatting issues and to proofread parts of the description. Part of my data processing code is adapted from the class example code from Dr.Fan.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
# hw3
library(Rtsne)
library(patchwork)
library(dplyr)
library(ggplot2)
library(gridExtra)
library(jcolors)
library(Seurat)
library(tidyverse)
library(viridis)
library(ggpubr)
setwd("/Users/tiya/Desktop/BME\ program\ info/Spring\ 2026/gemonic_data_visal")
## seperate data, normalize, PCA, tSNE, Kmeans, test the most upregulated genes in each cluster, use wilcoxon or T test to do DGE, heatmap
#### data ####
dat = read_csv("data/Visium-IRI-ShamR_matrix.csv") %>%
data.frame()
colnames(dat) %>% head()
dim(dat) # [1] 1224 19468
genes = setdiff(colnames(dat), c("...1", "x", "y"))
length(genes) #[1] 19465
#sort
dat = dat %>%
mutate(cellnames = `...1`) %>%
select(-c(`...1`))
rownames(dat) = dat$cellnames
#new data
pos <- dat[,c('x', 'y')]
dim(pos) #[1] 1224 2
gexp <- dat[, genes]
dim(gexp) #[1] 1224 19465
# normalize
totgexp = rowSums(gexp)
mat <- log10(gexp/totgexp * 1e6 + 1)
# PCA
pcs <- prcomp(mat)
df <- data.frame(pcs$x, pos)
ggplot(df, aes(x=x, y=y, col=PC1)) + geom_point(cex=0.8)
plot(pcs$sdev[1:50]) #I would take a safe PC = 10
sort(abs(pcs$rotation[, 1]), decreasing = TRUE)[1:5] #Nccrp1, Ppp1r1b, Epcam, Slc12a1, Scin
sort(abs(pcs$rotation[, 2]), decreasing = TRUE)[1:5] #Serpina1f, Cyp7b1, Mep1b, Slc22a13, Irx1
pc = 10
#tSNE
set.seed(2026209)
tsne = Rtsne::Rtsne(pcs$x[, 1:pc], dims = 2, perplexity = 30)
emb = as.data.frame(tsne$Y)
#Kmeans
var = numeric(20)
for (k in 1:20) {
km_result = kmeans(pcs$x[, 1:pc], centers = k)
var[k] <- km_result$tot.withinss
}
plot(1:20, var, type = "b",
xlab = "Number of Clusters (k)",
ylab = "Within-cluster variance",
main = "Elbow Method")
clusters = as.factor(kmeans(pcs$x[,1:pc], centers=3)$cluster) #picked cluster = 3 as the slope gets flat near that point
df = cbind(pos, emb, data.frame(clusters), data.frame(pcs$x)[, 1:10], mat)
ggplot(df, aes(x=PC1, y=PC2, col=clusters)) + geom_point(cex=0.8)
ggplot(df, aes(x=x, y=y, col=clusters)) + geom_point(cex=0.8)
ggplot(df, aes(x=V1, y=V2, col=clusters)) + geom_point(cex=0.8)
#### DGE ####
ser = CreateSeuratObject(counts = t(gexp))
LayerData(ser, assay = "RNA", layer = "data") = t(mat)
ser[[]] = cbind(ser[[]], df)
Idents(ser) = ser$clusters
markers = FindAllMarkers(ser, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25) #by default, test.use = "wilcox"
top_markers = markers %>%
group_by(cluster) %>%
filter(avg_log2FC > 0.4 & pct.1 >= 0.25 & p_val_adj < 0.05) %>%
top_n(n = 5, wt = avg_log2FC)
DoHeatmap(object = ser,
features = top_markers$gene, slot = "data") +
scale_fill_viridis(option = "H")
#### put plot togethor ####
small_axes = theme(
axis.title = element_text(size = 8),
axis.text = element_text(size = 7),
plot.title = element_text(size = 10)
)
p1 = ggplot(df, aes(x=x, y=y, col=clusters)) +
geom_point(cex=0.3) +
theme_bw() +
coord_fixed() +
labs(title = "Spatial map:\nk-means clusters",
x = "x (spatial)", y = "y (spatial)", color = "Cluster") +
small_axes +
theme(plot.title = element_text(size = 8)) +
guides(color = "none")
p2 = ggplot(df, aes(x = V1, y = V2, col=clusters)) +
geom_point(cex=0.3) +
theme_bw() +
labs(title = "t-SNE",
x = "tSNE1", y = "tSNE2", color = "Cluster") +
small_axes +
guides(color = "none")
p3 = ggplot(df, aes(x = PC1, y = PC2, col=clusters)) +
geom_point(cex=0.3) +
theme_bw() +
labs(title = "PCA",
x = "PC1", y = "PC2", color = "Cluster") +
small_axes
p4 = DoHeatmap(object = ser,
features = top_markers$gene, slot = "data") +
scale_fill_viridis(option = "H", name = "Transformed\nexpression\nlog10(CPM+1)") +
guides(color = "none") +
labs(title = "Top marker genes per cluster\n(FindAllMarkers: Wilcoxon)") +
theme(plot.title = element_text(size = 8),
legend.title = element_text(size = 7))
p5 = ggplot(df, aes(x=x, y=y, color=Slc5a12)) +
geom_point(cex=0.3) +
theme_bw() +
NoLegend() +
scale_color_viridis(option = "H", name = "Transformed\nexpression\nlog10(CPM+1)") +
labs(title = "Spatial expression:\nSlc5a12",
x = "x (spatial)", y = "y (spatial)") +
small_axes +
coord_fixed() +
theme(plot.title = element_text(size = 8))
p6 = ggplot(df, aes(x=clusters, y = Slc5a12,
fill=clusters, color = clusters)) +
geom_violin() +
geom_boxplot(fill = "white", width = 0.1, color = "black") +
theme_bw() +
stat_compare_means(
comparisons = combn(levels(df$clusters), 2, simplify = FALSE),
method = "wilcox.test",
label = "p.signif" # or "p.format"
) +
theme(legend.position="none") +
labs(title = "Slc5a12 by cluster\n(pairwise Wilcoxon)",
x = "Cluster", y = "log10(CPM+1)") +
small_axes +
ylim(0, 4.5) +
theme(plot.title = element_text(size = 8))
p7 = ggplot(df, aes(x = PC1, y = PC2, col=Slc5a12)) +
geom_point(cex=0.3) +
theme_bw() +
scale_color_viridis(option = "H") +
small_axes +
guides(color = "none") +
labs(title = "Slc5a12 expression\non PCA plot",
x = "PC1", y = "PC2") +
theme(plot.title = element_text(size = 8))
p8 = ggplot(df, aes(x = V1, y = V2, col=Slc5a12)) +
geom_point(cex=0.3) +
theme_bw() +
scale_color_viridis(option = "H") +
small_axes +
guides(color = "none") +
labs(title = "Slc5a12 expression\n on t-SNE plot",
x = "tSNE1", y = "tSNE2") +
theme(plot.title = element_text(size = 8))
#verification of wilcoxon test results
x1 = df %>% filter(clusters == "2") %>% pull(Slc5a12)
x2 = df %>% filter(clusters == "1") %>% pull(Slc5a12)
wilcox.test(x1, x2, alternative='greater') #for 2 vs 3, W = 104426, p-value < 2.2e-16
# for 1 vs 3, W = 24664, p-value = 0.006351
# for 2 vs 1, W = 173794, p-value < 2.2e-16
## put togethor
top <- (p1 | p2 | p3) +
plot_layout( widths = c(1, 1, 1, 0.2)) &
plot_annotation(theme = theme(legend.position = "right"))
bottom <- (((p6 / p5) | (p7 / p8)) | p4)+
plot_layout(widths = c(1, 1, 1.5, 0.2)) &
plot_annotation(theme = theme(legend.position = "right"))
(top / bottom) +
plot_layout(heights = c(1, 3)) +
plot_annotation(tag_levels = "A")