Animated Comparison of Normalized vs. Non-Normalized Acox2 Expression

Description
In HW2, I had forgotten to normalize my data by library size. So, when seeing “What happens if I do or not not normalize and/or transform the gene expression data (e.g. log and/or scale) prior to dimensionality reduction?” As an exploratory option for EC1, I thought it would be interesting to see what would have happened if I had not learned from my mistake in HW2 and had also failed to normalize the gene expression data when I approached HW3.
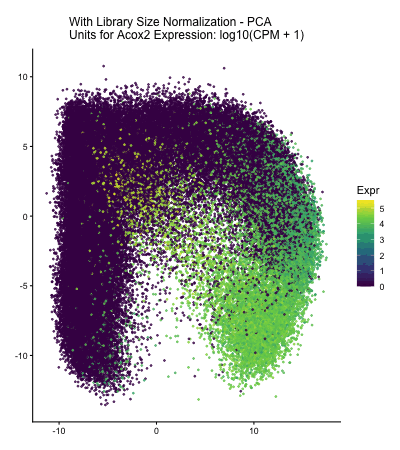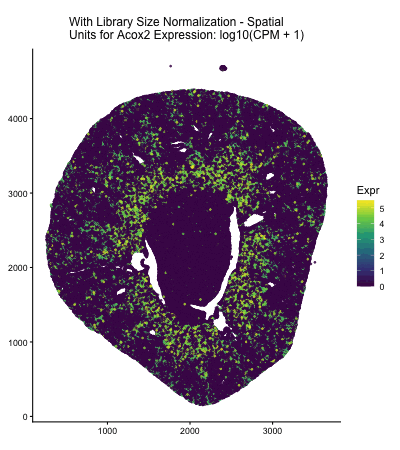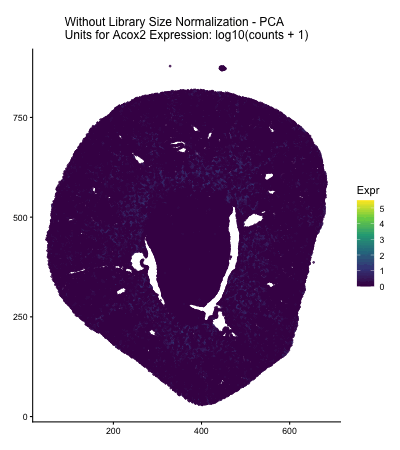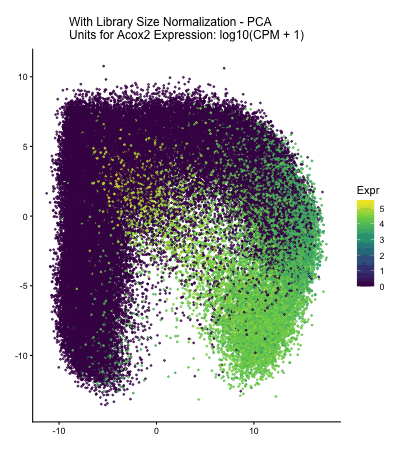
The data visualization cycles between four major visualizations derived from HW3. In HW3, we were asked to characterize a cell type for a cluster in the Xenium dataset, and in this assignment Acox2 upregulation in my cluster of interest played a large role in my correct characterization of my cluster of interest as proximal tubule cells. In the PCA and spatial plots, we see areas of high Acox2 expression (encoded by the lighter color saturation), in the areas that corresponded with the cluster of interest in HW3&4. Meanwhile, when we fail to normalize the data prior to our dimensionality reduction, we obtain a squished PCA plot and almost no Acox2 expression. This seems to indicate that if I had made the same mistake and failed to normalize the gene expression data prior to my dimensionality reduction and clustering analysis, I would have failed to identify the Acox2 upregulation that led me to correctly identify the cell type of the cluster of interest in HW3.
Code
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
# giving Claude access to the animation we made in class as well as my submission
# for HW3, I asked Claude to generate the R code for an animation that switches between
# the PCA and spatial expression graphs for Acox2, the genetic marker for Proximal Tubule Cells
# with and without normalization (CPM), seeing if failing to normalize the data would have
# affected my conclusions in HW3
# import libraries
library(ggplot2)
library(gganimate)
library(dplyr)
library(magick)
# read in the data
data <- read.csv('/Users/henryaceves/Desktop/JHU/S2/GDV/GDV datasets/Xenium-IRI-ShamR_matrix.csv.gz')
# build the dataframe
pos <- data[,c('x','y')]
rownames(pos) <- data[,1]
gexp <- data[,4:ncol(data)]
rownames(gexp) <- data[,1]
#---------------------------------------------------------
# TWO PARALLEL PIPELINES: Normalized vs Non-Normalized
#---------------------------------------------------------
# === NORMALIZED PIPELINE (CPM + log) ===
totgexp <- rowSums(gexp)
mat_norm <- log10(gexp / totgexp * 1e6 + 1)
pcs_norm <- prcomp(mat_norm, center = TRUE, scale = FALSE)
toppcs_norm <- pcs_norm$x[, 1:10]
set.seed(123)
clusters_norm <- as.factor(kmeans(toppcs_norm, centers = 9, nstart = 25)$cluster)
# === NON-NORMALIZED PIPELINE (just log, no CPM) ===
mat_raw <- log10(gexp + 1) # log transform only, no library size normalization
pcs_raw <- prcomp(mat_raw, center = TRUE, scale = FALSE)
toppcs_raw <- pcs_raw$x[, 1:10]
set.seed(123)
clusters_raw <- as.factor(kmeans(toppcs_raw, centers = 9, nstart = 25)$cluster)
#---------------------------------------------------------
# CREATE ANIMATION DATAFRAMES
#---------------------------------------------------------
# State 1: Normalized data - PCA space
df1 <- data.frame(
x_plot = pcs_norm$x[, "PC1"],
y_plot = pcs_norm$x[, "PC2"],
gene = mat_norm[, "Acox2"],
state = "Normalized (CPM) - PCA"
)
# State 2: Normalized data - Spatial space
df2 <- data.frame(
x_plot = pos$x,
y_plot = pos$y,
gene = mat_norm[, "Acox2"],
state = "Normalized (CPM) - Spatial"
)
# State 3: Non-normalized data - PCA space
df3 <- data.frame(
x_plot = pcs_raw$x[, "PC1"],
y_plot = pcs_raw$x[, "PC2"],
gene = mat_raw[, "Acox2"],
state = "Non-Normalized - PCA"
)
# State 4: Non-normalized data - Spatial space
df4 <- data.frame(
x_plot = pos$x,
y_plot = pos$y,
gene = mat_raw[, "Acox2"],
state = "Non-Normalized - Spatial"
)
# Combine all states
df_combined <- bind_rows(df1, df2, df3, df4)
# Set state order for animation sequence
df_combined$state <- factor(df_combined$state,
levels = c("Normalized (CPM) - PCA",
"Normalized (CPM) - Spatial",
"Non-Normalized - PCA",
"Non-Normalized - Spatial"))
#---------------------------------------------------------
# CREATE ANIMATION
#---------------------------------------------------------
anim <- ggplot(df_combined, aes(x = x_plot, y = y_plot, col = gene)) +
geom_point(size = 0.5, alpha = 0.7) +
scale_color_viridis_c() +
transition_states(state,
transition_length = 2,
state_length = 2) +
labs(title = '{closest_state}',
subtitle = "Acox2 Expression",
x = NULL,
y = NULL,
col = "Expr") +
ease_aes('cubic-in-out') +
view_follow(fixed_y = FALSE, fixed_x = FALSE) +
theme_classic() +
theme(
plot.title = element_text(hjust = 0.5, face = "bold", size = 14),
plot.subtitle = element_text(hjust = 0.5, size = 12),
axis.text = element_text(color = "black")
)
# Render animation
animate(anim, nframes = 200, fps = 10, width = 400, height = 400,
renderer = magick_renderer())
# Save if desired
# anim_save("normalization_comparison.gif")