HW EC1: PC1 Loadings of Top 5 Log-Normalized Genes Across PCA Preprocessing Methods

Description
Question: What happens if I do or not not normalize and/or transform the gene expression data (e.g. log and/or scale) prior to dimensionality reduction?
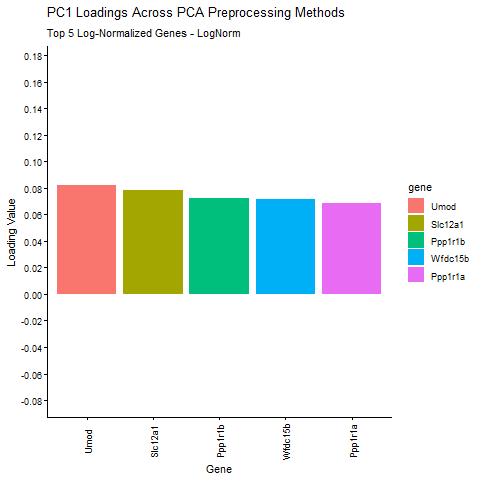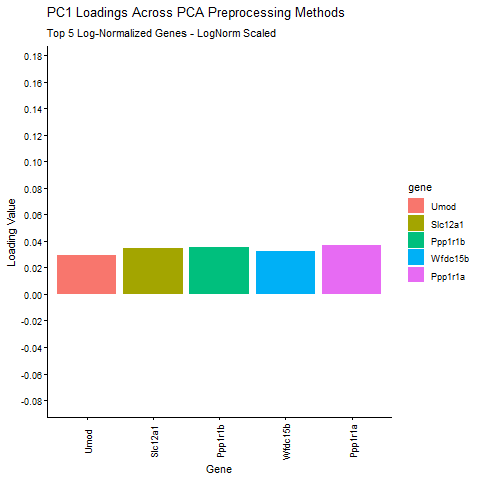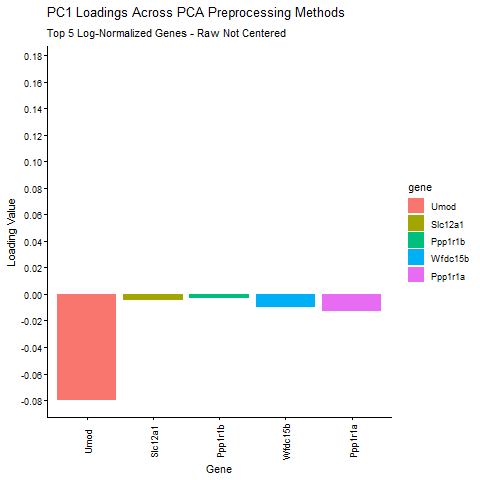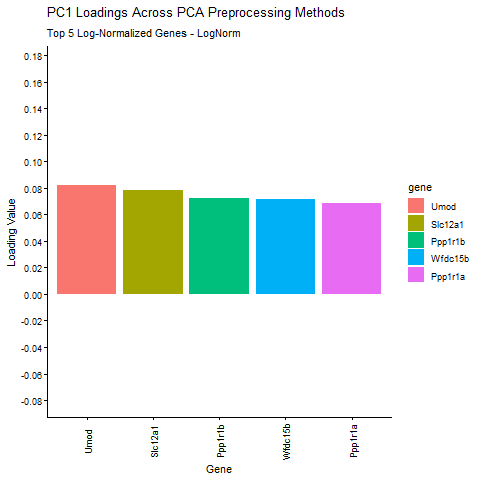
I am exploring how different preprocessing choices affect the structure and interpretability of principal component analysis (PCA) in spatial transcriptomics data. Starting from raw Visium gene expression counts, the pipeline applies library‑size normalization and log transformation, filters out zero‑variance genes, and then computes six PCA variants that isolate the effects of centering, scaling, and log transformation. I am comparing how library size log transformation affects the loading values, and how changing the PCA parameters (Centering and scaling) affects the PCA loadings. To make the comparison biologically meaningful, the top five PC1 genes from the log‑normalized PCA (the most informative genes under proper normalization taught in class) are identified and their PC1 loadings are extracted from every PCA variant. The animated barplot visualizes how the loadings of these same five genes change across preprocessing methods. The animimation shows that raw PCA is dominated by technical effects such as library size (the cell density of the spot are affecting the loading values), scaling reshapes the variance structure and can shrink biologically relevant loadings (makes them look uniform. In ST, we want to keep variance because it is usually biologically meaningful, ans scaling removes the signal), and omitting centering makes PC1 align toward the mean expression vector instead of the variation around the mean. Skipping centering can change the direction of the PC1 axis, which can cause the entire loading vector to flip signs, which is why some of the loading values change from positive to negative. In contrast, log‑normalized PCA produces stable, interpretable loadings that reflect underlying biology. These results show that normalization, log transformation, and centering are important for PCA to capture meaningful biological variation rather than technical artifacts.
Code
library(patchwork)
library(ggplot2)
library(gganimate)
data <- read.csv('C:/users/lilli/Downloads/Visium-IRI-ShamR_matrix.csv.gz')
#gene expression data
gexp <- data[, 4:ncol(data)]
#rownames are the spots
rownames(gexp) <- data[,1]
#positions of the data
pos <- data[, c('x', 'y')]
#library size normalization
total_genexp<- rowSums(gexp)
log_norm_gexp<- log10(gexp/total_genexp*1e4+1)
#remove genes with zero variance
gexp<- gexp[, apply(gexp, 2, var) > 0]
log_norm_gexp<- log_norm_gexp[, apply(log_norm_gexp, 2, var) > 0]
#PCA variants
#raw counts
pca_raw <- prcomp(gexp)
#scaled raw
pca_raw_scale <- prcomp(gexp, center= TRUE, scale=TRUE)
pca_raw_nocenter <- prcomp(gexp, center=FALSE, scale= FALSE)
#library_size norm
pca_log<- prcomp(log_norm_gexp)
pca_log_scale<- prcomp(log_norm_gexp, center= TRUE, scale=TRUE)
pca_log_nocenter<- prcomp(log_norm_gexp, center=FALSE, scale= FALSE)
#get the top 5 genes in the log-norm pca
top5_log<- sort(pca_log$rotation[,1], decreasing = TRUE)[1:5]
top5_genes<- names(top5_log)
#function to extract top 5 PC1 loadings for each PCA method
extract_top<- function(pca, label, genes) {
df<- data.frame(gene= genes,
loading= pca$rotation[genes, 1],
state= label)
df$gene <- factor(df$gene, levels = genes)}
#build dataframe for all 6 PCA variants (learned this form HW4)
loading_df <- rbind(
extract_top(pca_raw, 'Raw', top5_genes),
extract_top(pca_raw_scale, 'Raw Scaled', top5_genes),
extract_top(pca_raw_nocenter, 'Raw Not Centered', top5_genes),
extract_top(pca_log, 'LogNorm', top5_genes),
extract_top(pca_log_scale, 'LogNorm Scaled', top5_genes),
extract_top(pca_log_nocenter, 'LogNorm Not Centered', top5_genes))
#I followed this tutorial: https://r-graph-gallery.com/package/gganimate.html
#I used this for some of the choices into the animation
#https://rstudio.github.io/cheatsheets/gganimate.pdf
anim_plot <- ggplot(loading_df, aes(x = gene, y = loading, fill = gene)) +
geom_bar(stat = 'identity') +
theme_classic() +
theme(axis.text.x = element_text(angle = 90, vjust = 0.5,))+
transition_states(state, transition_length = 2, state_length = 1) +
#Asked AI: How to add more ticks to my y-axis?
scale_y_continuous(breaks = seq(-0.2, 0.2, by = 0.02))+
ease_aes('cubic-in-out') +
xlab('Gene') +
ylab('Loading Value') +
#asked AI: how to change the title of each image in gganimate?
labs(title='PC1 Loadings Across PCA Preprocessing Methods', subtitle = 'Top 5 Log-Normalized Genes - {closest_state}')
#asked AI: how to slow down gganimate object?
animate(anim_plot, fps = 5)
anim_save('llam9.gif')