Running tSNE analysis on genes or PCs

What data types are you visualizing?
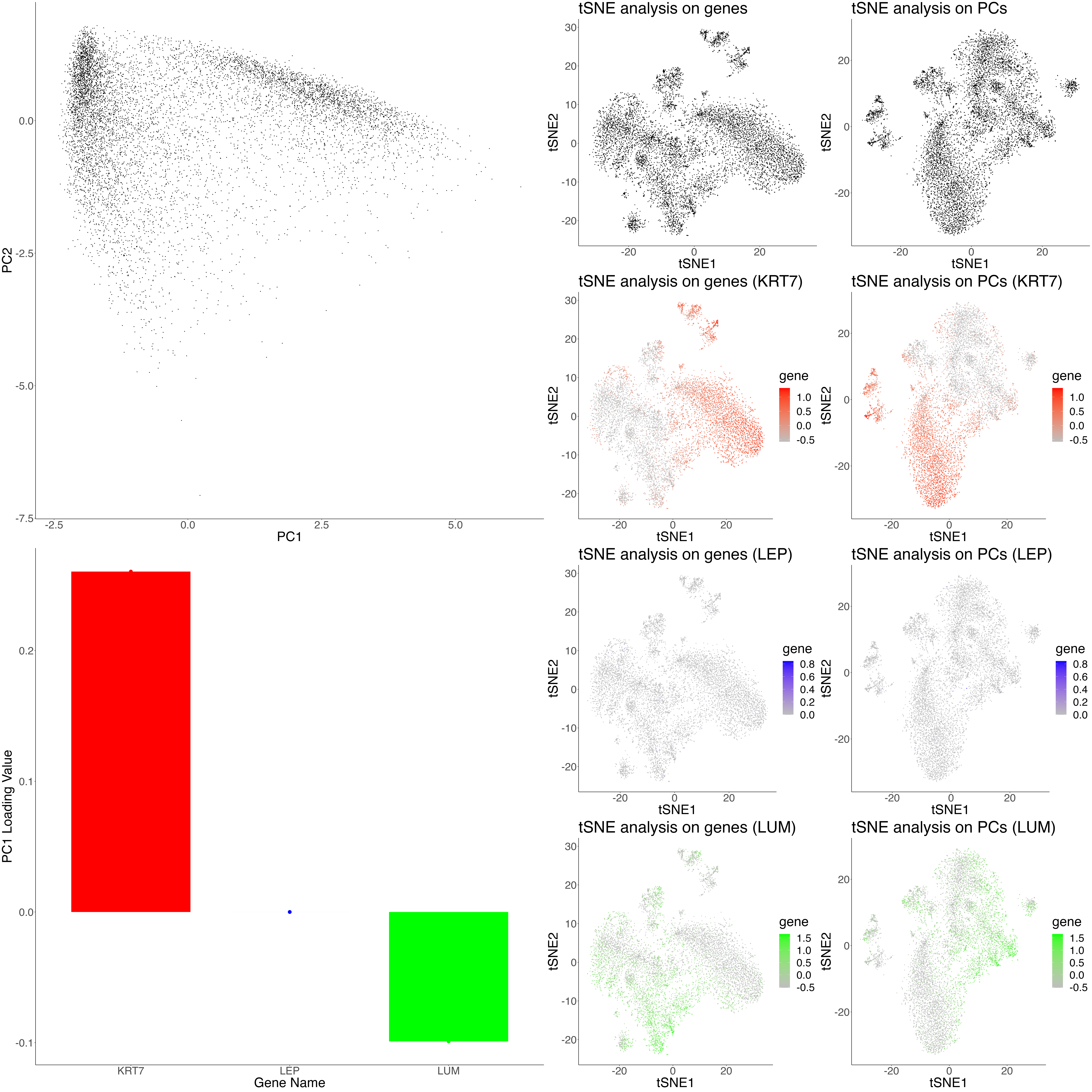
In this multi-panel plot, I am visualizing various quantitative and categorical data. For the PCA plot on the upper left, I am visualizing quantitative data of each cell’s position in the transformed space based on principle component 1 (PC1) and PC2 axes. For the loading plot on the lower left, I am visualizing quantitative data of PC1 loading value for specific genes and categorical data of three genes, KRT7, LEP, and LUM. For the tSNE plots on the right, I am visualizing quantitative data of each cell’s position in the transformed space based on tSNE1 and tSNE2 axes. For tSNE plots that are colored, I am also visualizing quantitative data of each cell’s mean-centered log-scaled expression of a chosen gene. Depending on which tSNE plots you look at, I am visualizing the same categorical data of three genes as the loading plot.
What data encodings are you using to visualize these data types?
For the PCA plot on the upper left, each cell is represented as geometric primitive of points. To encode each cell’s position in the transformed space based on PC1 and PC2 axes, I used the visual channel of position along the x axis and y axis for PC1 and PC2, respectively. For the loading plot on the lower left, each gene’s PC1 loading value is represented as both geometric primitive of points and lines because it was difficult to see LEP with only geometric primitive of lines. To encode each gene’s loading value, I am using the visual channel of position so that each bar starts at loading value = 0 and size of the lines. To encode categorical data of gene, I am using the visual channel of color hue so that KRT7, LEP, and LUM are represented as red, blue, and green. For the tSNE plots on the right, each cell is represented as geometric primitive of points. To encode each cell’s position in the transformed space based on tSNE1 and tSNE2 axes, I used the visual channel of position along the x axis and y axis for tSNE1 and tSNE2, respectively. To encode each cell’s mean-centered log-scaled expression of a chosen gene, I am using the visual channel color saturation. To encode which gene the tSNE plots are showing, I am using the visual channel of color hue so that KRT7, LEP, and LUM are represented as red, blue, and green.
What type of data visualization is this? What about the data are you trying to make salient through this data visualization?
In this multi-panel plot, I seek to make more salient whether we should run tSNE analysis on genes or PCs. In order to generate the plot, I log scaled and mean-centered the raw data. Then, I run PCA on this normalized data. tSNE analysis was run on both the normalized data (denoted as genes on plots) and the result of PCA analysis of the normalized data, which is essentially the normalized data transformed to PCs space (denoted as PCs on plots). In order to compare tSNE analysis on genes and PCs, I decided to compare the expression of genes with three different PC1 loading values. With the highest loading value, KRT7 is the gene most positively affected by PC1. With the lowest positive loading value, LEP is the gene least positively affected by PC1. With the lowest loading value, LUM is the gene most negatively affected by PC1. Plots on the left show the results of PCA analysis, and plots on the right show the results of tSNE analysis. Left column of tSNE plots show tSNE analysis on genes, and right column of tSNE plots show tSNE analysis on PCs. As it can be seen by the general shape of tSNE plots, tSNE analysis on genes and PCs generate extremely similar shape but rotated and flipped around a certain axis. In addition, gene expression of KRT7, LEP, and LUM all show same distribution along the tSNE plot shapes. Therefore, it can be concluded that tSNE analysis on genes and PCs generate very similar results, and it is not required to run PCA prior to tSNE.
What Gestalt principles or knowledge about perceptiveness of visual encodings are you using to accomplish this?
I am using the Gestalt principle of similarity to indicate that red, blue, and green represent data related to KRT7, LEP, and LUM. This would allow the audience to easily associate the loading values and the tSNE plots of each gene. I am also using the Gestalt principle of similarity to group different types of plots (PCA plot, loading plot, and tSNE plots). Based on the literature on human perception processing different encoding for different types, position and length best represent quantitative data, and position and hue best represent categorical data. This knowledge was applied to improve the saliency of the plots.
Code
setwd('~/Desktop/Johns Hopkins University/4 - Senior (2022 - 2023)/Spring/Genomic Data Visualization/HW/HW3')
getwd()
## load library
library(ggplot2)
library(Rtsne)
library(gridExtra)
## set seed for reproducibility
set.seed(0)
## set custom theme for plots
plot_theme <- theme_classic() +
theme(
text = element_text(size = 35),
legend.key.size = unit(1, 'cm')
)
## read in data
data <- read.csv('~/Desktop/genomic data visualization 2023 data (copy)/bulbasaur.csv.gz', row.names = 1)
# extract gene expression
gexp <- data[,4:ncol(data)]
# log scale
mat <- log10(gexp+1)
## check mean and variance for each gene
head(sort(apply(mat, 2, mean), decreasing = TRUE))
head(sort(apply(mat, 2, var), decreasing = TRUE))
## normalization using scale (set mean = 0, variance = 1)
nor <- scale(mat, center=TRUE, scale=FALSE)
head(sort(apply(nor, 2, mean), decreasing = TRUE))
head(sort(apply(nor, 2, var), decreasing = TRUE))
## pca analysis
pcs <- prcomp(nor)
# plot all
df_pca_nor <- data.frame(pcs$x[,1:2])
ggplot(data = df_pca_nor, aes(x = PC1, y = PC2)) +
geom_point(size = 0.3) +
theme_classic()
df_pca_nor <- data.frame(pcs$x[,1:2])
ppca <- ggplot(data = df_pca_nor, aes(x = PC1, y = PC2)) +
geom_point(size = 0.3) +
plot_theme
# look at loadings for both PC1 and PC2
# PC1
# positively affected
head(sort(pcs$rotation[,1], decreasing=TRUE))
# negatively affected
head(sort(pcs$rotation[,1], decreasing=FALSE))
# PC2
# positively affected
head(sort(pcs$rotation[,2], decreasing=TRUE))
# negatively affected
head(sort(pcs$rotation[,2], decreasing=FALSE))
# plot loading values for specific genes
gene_loading <- data.frame(PC1 = pcs$rotation[,1])
df_loading <- data.frame(
gene_name = c('KRT7', 'LEP', 'LUM'),
loading = c(gene_loading['KRT7',], gene_loading['LEP',],gene_loading['LUM',])
)
ploading <- ggplot(data = df_loading, aes(x = gene_name, y = loading, fill = gene_name)) +
geom_bar(stat = 'identity', width = 0.75) +
scale_fill_manual(values = c('red','blue','green')) +
geom_point(aes(col = gene_name), size = 4) +
scale_color_manual(values = c('red','blue','green')) +
xlab('Gene Name') +
ylab('PC1 Loading Value') +
plot_theme +
theme(legend.position = 'none')
# plot specific gene
df_pca_nor <- data.frame(pcs$x[,1:2], gene=nor[,'FOXC2'])
ggplot(data = df_pca_nor, aes(x = PC1, y = PC2, col = gene)) +
geom_point(size = 0.3) +
scale_color_gradient(low='grey', high='red') +
theme_classic()
## tSNE analysis on genes
tsne_genes <- Rtsne(
nor,
pca = FALSE,
pca_center = FALSE,
pca_scale = FALSE)
df_tsne_nor <- data.frame(tsne_genes$Y)
# plot all
p1 <- ggplot(data = df_tsne_nor, aes(x = X1, y = X2)) +
geom_point(size = 0.3) +
xlab('tSNE1') +
ylab('tSNE2') +
ggtitle('tSNE analysis on genes') +
plot_theme
# plot LEP (least positively affected)
df_tsne_nor_gene <- data.frame(tsne_genes$Y, gene=nor[,'LEP'])
p3 <- ggplot(data = df_tsne_nor_gene, aes(x = X1, y = X2, col = gene)) +
geom_point(size = 0.3) +
scale_color_gradient(low='grey', high='blue') +
xlab('tSNE1') +
ylab('tSNE2') +
ggtitle('tSNE analysis on genes (LEP)') +
plot_theme
# plot KRT7 (most positively affected)
df_tsne_nor_gene <- data.frame(tsne_genes$Y, gene=nor[,'KRT7'])
p5 <- ggplot(data = df_tsne_nor_gene, aes(x = X1, y = X2, col = gene)) +
geom_point(size = 0.3) +
scale_color_gradient(low='grey', high='red') +
xlab('tSNE1') +
ylab('tSNE2') +
ggtitle('tSNE analysis on genes (KRT7)') +
plot_theme
# plot LUM (most negatively affected)
df_tsne_nor_gene <- data.frame(tsne_genes$Y, gene=nor[,'LUM'])
p7 <- ggplot(data = df_tsne_nor_gene, aes(x = X1, y = X2, col = gene)) +
geom_point(size = 0.3) +
scale_color_gradient(low='grey', high='green') +
xlab('tSNE1') +
ylab('tSNE2') +
ggtitle('tSNE analysis on genes (LUM)') +
plot_theme
## tSNE analysis on PCs
df_pca_nor_allpc <- data.frame(pcs$x)
tsne_pc <- Rtsne(
df_pca_nor_allpc,
pca = FALSE,
pca_center = FALSE,
pca_scale = FALSE)
# plot all
df_tsne_pc <- data.frame(tsne_pc$Y)
p2 <- ggplot(data = df_tsne_pc, aes(x = X1, y = X2)) +
geom_point(size = 0.3) +
xlab('tSNE1') +
ylab('tSNE2') +
ggtitle('tSNE analysis on PCs') +
plot_theme
# plot LEP (least positively affected)
df_tsne_pc_gene <- data.frame(tsne_pc$Y, gene=nor[,'LEP'])
p4 <- ggplot(data = df_tsne_pc_gene, aes(x = X1, y = X2, col = gene)) +
geom_point(size = 0.3) +
scale_color_gradient(low='grey', high='blue') +
xlab('tSNE1') +
ylab('tSNE2') +
ggtitle('tSNE analysis on PCs (LEP)') +
plot_theme
# plot KRT7 (most positively affected)
df_tsne_pc_gene <- data.frame(tsne_pc$Y, gene=nor[,'KRT7'])
p6 <- ggplot(data = df_tsne_pc_gene, aes(x = X1, y = X2, col = gene)) +
geom_point(size = 0.3) +
scale_color_gradient(low='grey', high='red') +
xlab('tSNE1') +
ylab('tSNE2') +
ggtitle('tSNE analysis on PCs (KRT7)') +
plot_theme
# plot LUM (most negatively affected)
df_tsne_pc_gene <- data.frame(tsne_pc$Y, gene=nor[,'LUM'])
p8 <- ggplot(data = df_tsne_pc_gene, aes(x = X1, y = X2, col = gene)) +
geom_point(size = 0.3) +
scale_color_gradient(low='grey', high='green') +
xlab('tSNE1') +
ylab('tSNE2') +
ggtitle('tSNE analysis on PCs (LUM)') +
plot_theme
lay <- rbind(c(1,1,3,4),
c(1,1,5,6),
c(2,2,7,8),
c(2,2,9,10))
grid.arrange(ppca, ploading, p1, p2, p5, p6, p3, p4, p7, p8, layout_matrix = lay)
result <- grid.arrange(ppca, ploading, p1, p2, p5, p6, p3, p4, p7, p8, layout_matrix = lay)
## save plot
ggsave("hw3_gaihara1.png", plot = result, width = 40, height = 40, dpi = 300)
## references
# whether it is important to center/scale one's data prior to tSNE analysis: https://stats.stackexchange.com/questions/164917/should-data-be-centeredscaled-before-applying-t-sne
# whether it is important to center one's data prior to PCR analysis using prcomp: https://stats.stackexchange.com/questions/189822/how-does-centering-make-a-difference-in-pca-for-svd-and-eigen-decomposition
# ggplot2 main, axis, and legend titles: http://www.sthda.com/english/wiki/ggplot2-title-main-axis-and-legend-titles
# arranging multiple grobs on a page: https://cran.r-project.org/web/packages/gridExtra/vignettes/arrangeGrob.html
# setting custom theme in ggplot: http://www.sthda.com/english/wiki/ggplot2-themes-and-background-colors-the-3-elements
# change font size of ggplot: https://statisticsglobe.com/change-font-size-of-ggplot2-plot-in-r-axis-text-main-title-legend
# change legend size: https://www.statology.org/ggplot2-legend-size/
# barplot with ggplot: https://r-graph-gallery.com/218-basic-barplots-with-ggplot2.html
# manually change color in ggplot: https://www.geeksforgeeks.org/change-color-of-bars-in-barchart-using-ggplot2-in-r/
# remove legend in ggplot: https://statisticsglobe.com/remove-legend-ggplot2-r#:~:text=Example%201%3A%20Remove%20All%20Legends%20in%20ggplot2&text=We%20simply%20had%20to%20specify,get%20rid%20of%20both%20legends.
(do you think my data visualization is effective? why or why not?)