Reference-free cell-type deconvolution of multi-cellular spatially resolved transcriptomics data
STdeconvolve
![]()
STdeconvolve enables reference-free cell-type deconvolution of multi-cellular pixel-resolution spatially resolved transcriptomics data
![]()
The overall approach is now published in Nature Communications
Overview
STdeconvolve is an unsupervised machine learning approach to deconvolve multi-cellular pixel-resolution spatial transcriptomics datasets in order to recover the putative transcriptomic profiles of cell-types and their proportional representation within spatially resolved pixels without reliance on external single-cell transcriptomics references.

Example
library(STdeconvolve)
## load built in data
data(mOB)
pos <- mOB$pos
cd <- mOB$counts
annot <- mOB$annot
## remove pixels with too few genes
counts <- cleanCounts(cd, min.lib.size = 100)
## feature select for genes
corpus <- restrictCorpus(counts, removeAbove=1.0, removeBelow = 0.05)
## choose optimal number of cell-types
ldas <- fitLDA(t(as.matrix(corpus)), Ks = seq(2, 9, by = 1))
## get best model results
optLDA <- optimalModel(models = ldas, opt = "min")
## extract deconvolved cell-type proportions (theta) and transcriptional profiles (beta)
results <- getBetaTheta(optLDA, perc.filt = 0.05, betaScale = 1000)
deconProp <- results$theta
deconGexp <- results$beta
## visualize deconvolved cell-type proportions
vizAllTopics(deconProp, pos,
groups = annot,
group_cols = rainbow(length(levels(annot))),
r=0.4)
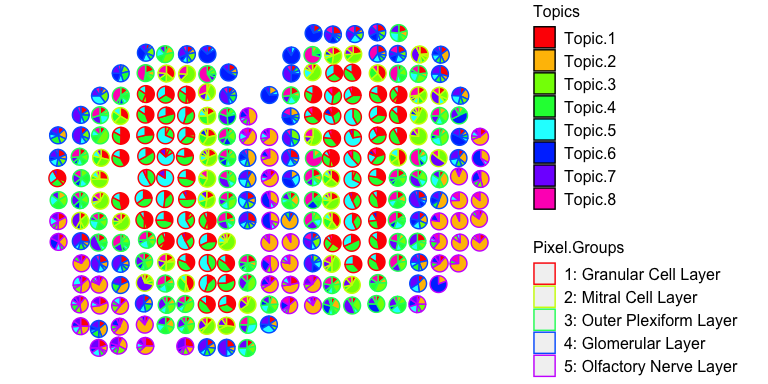
More details can be found in the tutorials.
Tutorials
- Getting started with
STdeconvolve - Additional features with
STdeconvolve - Annotating deconvolved cell-types
- Analysis of 10X Visium data
- Examples of when
STdeconvolvemay fail - Discussion and examples of processing multiple datasets together or separately
Installation
To install STdeconvolve, we recommend using remotes:
require(remotes)
remotes::install_github('JEFworks-Lab/STdeconvolve')
STdeconvolve is also now available through Bioconductor.
Note that through Bioconductor (release 3.15), the R version must be >=4.2.
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
# The following initializes usage of Bioc devel
BiocManager::install(version='devel')
BiocManager::install("STdeconvolve")
Contributing
We welcome any bug reports, enhancement requests, general questions, and other contributions. To submit a bug report or enhancement request, please use the STdeconvolve GitHub issues tracker. For more substantial contributions, please fork this repo, push your changes to your fork, and submit a pull request with a good commit message.
Reproducing Analyses
Links below point to code outlining the preprocessing of datasets used for analyses in the manuscript.
- MERFISH mouse medial preoptic area (Moffit et al. 2018)
- Mouse Olfactory Bulb (Stahl et al. 2016)
- Breast cancer sections (Yoosuf et al. 2020)
- DBiT-seq of E11 mouse lower embryo and tail (Liu et al. 2020)
Examples of STdeconvolve being used in “the wild”
We are extremely excited and humbled that STdeconvolve is being applied by members of the scientific community! STdeconvolve is incorporated into 10X Genomics spaceranger v2.1 spot deconvolution pipeline!.
Citing STdeconvolve
If you use STdeconvolve in your research paper, please cite:
Miller BF, Huang F, Atta L, Sahoo A, Fan J. Reference-free cell type deconvolution of multi-cellular pixel-resolution spatially resolved transcriptomics data. Nat Commun. 2022 Apr 29;13(1):2339.
Similarly, if you use the reference-free spot deconvolution results from Space Ranger v2.1 and later, please cite to refer readers to our original work demonstrating its utility and application to multi-cellular pixel-resolution spatial transcriptomics data.