Major Publications
Find our major scientific contributions on this page. For a full list of all associated publications, please see ORCiD or Google Scholar. We are committed to open access. If you find a manuscript that we have co-authored but is not available here, please contact us directly and we will be happy to send you an electronic copy.
* Denotes equal contribution
^ Denotes corresponding author
JEFworks lab members are underlined
Table of Contents
- 02 July 2026 - Another 10 years of PLOS Computational Biology - A data-driven reflection on trends in genomics research
- 01 July 2026 - Four-dimensional molecular mapping from a spatial snapshot reveals the dynamics of hair follicle organogenesis
- 20 June 2026 - Spatiotemporal transcriptomic analysis during cold ischemic injury to the murine kidney reveals compartment-specific changes
- 18 December 2025 - Spatial Transcriptomics As Rasterized Image Tensors (STARIT) characterizes cell states with subcellular molecular heterogeneity
- 21 November 2025 - STcompare - comparative spatial transcriptomics data analysis of structurally matched tissues to characterize differentially spatially patterned genes
- 03 September 2025 - Impact of Data Quality on Deep Learning Prediction of Spatial Transcriptomics from Histology Images
- 31 March 2025 - Evidence of off-target probe binding in the 10x Genomics Xenium v1 Human Breast Gene Expression Panel compromises accuracy of spatial transcriptomic profiling
- 31 January 2025 - scatterbar - an R package for visualizing proportional data across spatially resolved coordinates
- 03 January 2025 - Characterizing cell-type spatial relationships across length scales in spatially resolved omics data
- 20 June 2024 - SEraster - a rasterization preprocessing framework for scalable spatial omics data analysis
- 12 June 2024 - Gene count normalization in single-cell imaging-based spatially resolved transcriptomics
- 16 May 2024 - Single-cell morphology encodes functional subtypes of senescence in aging human dermal fibroblasts
- 08 December 2023 - STalign - alignment of spatial transcriptomics data using diffeomorphic metric mapping
- 07 December 2023 - Soluble PD-L1 reprograms blood monocytes to prevent cerebral edema and facilitate recovery after ischemic stroke
- 28 November 2023 - Single cell and spatial transcriptomics analysis of kidney double negative T lymphocytes in normal and ischemic mouse kidneys
- 27 April 2023 - Why it's worth making computational methods easy to use
- 29 March 2023 - Cross-modality mapping using image varifolds to align tissue-scale atlases to molecular-scale measures with application to 2D brain sections
- 29 April 2022 - Reference-free cell type deconvolution of pixel-resolution spatially resolved transcriptomics data
- 12 January 2022 - Single cell analysis reveals immune dysfunction from the earliest stages of CLL that can be reversed by ibrutinib
- 28 September 2021 - VeloViz - RNA-velocity informed embeddings for visualizing cellular trajectories
- 06 September 2021 - Computational challenges and opportunities in spatially resolved transcriptomic data analysis
- 03 September 2021 - Rewiring of human neurodevelopmental gene regulatory programs by human accelerated regions
- 25 June 2021 - Multi Scale Diffeomorphic Metric Mapping of Spatial Transcriptomics Datasets
- 03 June 2021 - Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma
- 25 May 2021 - Characterizing spatial gene expression heterogeneity in spatially resolved single-cell transcriptomics data with nonuniform cellular densities
- 08 December 2020 - Spatial organization of the transcriptome in individual neurons
- 15 September 2020 - Single-cell transcriptomics in cancer - computational challenges and opportunities
- 19 November 2019 - Fast, sensitive and accurate integration of single-cell data with Harmony
- 09 September 2019 - Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression
- 11 February 2019 - A Murine Model of Chronic Lymphocytic Leukemia Based on B Cell-Restricted Expression of Sf3b1 Mutation and Atm Deletion
- 08 August 2018 - RNA velocity of single cells
- 13 June 2018 - Linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell RNA-seq data
- 11 December 2017 - Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain
- 22 May 2017 - Integrated single-cell genetic and transcriptional analysis suggests novel drivers of chronic lymphocytic leukemia
- 03 November 2016 - Transcriptomic Characterization of SF3B1 Mutation Reveals Its Pleiotropic Effects in Chronic Lymphocytic Leukemia
- 25 August 2016 - Cell-Type-Specific Alternative Splicing Governs Cell Fate in the Developing Cerebral Cortex
- 20 May 2016 - Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition
- 18 January 2016 - Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis
- 08 December 2014 - Locally Disordered Methylation Forms the Basis of Intratumor Methylome Variation in Chronic Lymphocytic Leukemia
Another 10 years of PLOS Computational Biology - A data-driven reflection on trends in genomics research
Jean Fan
Paper: PLOS Computational Biology. July 2, 2026. doi.org/10.1371/journal.pcbi.1014471 | Pubmed
Four-dimensional molecular mapping from a spatial snapshot reveals the dynamics of hair follicle organogenesis
Soichiro Asami, Chenshuo Yin, Jean Fan, Luis A. Garza, Reza Kalhor
Paper: Cell. July 01, 2026. doi.org/10.1016/j.cell.2026.06.014 | Pubmed
Spatiotemporal transcriptomic analysis during cold ischemic injury to the murine kidney reveals compartment-specific changes
Srujan Singh, Shishir Kumar Patel, Ryo Matsuura, Dee Velazquez, Zhaoli Sun, Sanjeev Noel, Hamid Rabb^, Jean Fan^
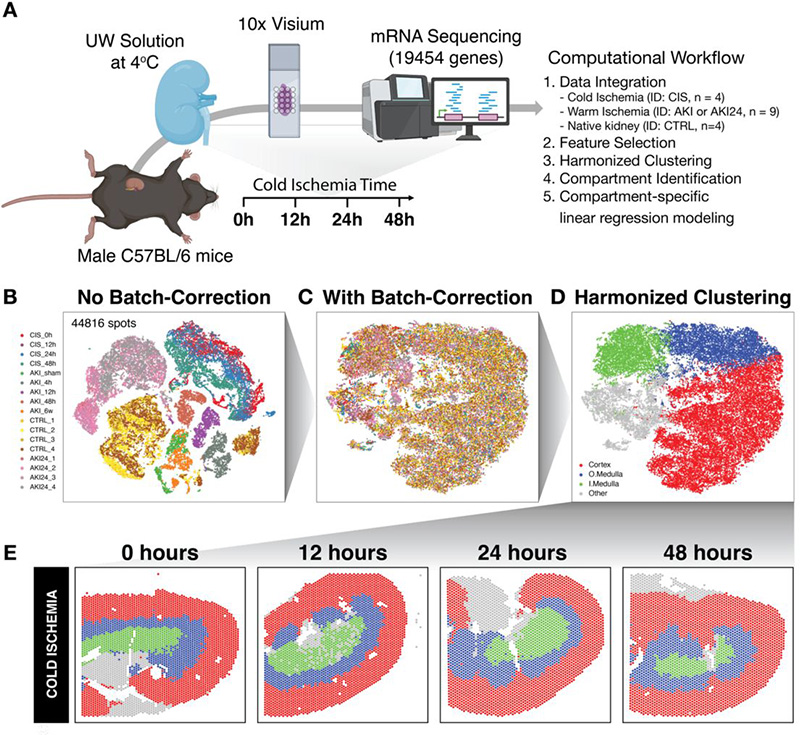
Paper: Genome Biology. June 20, 2026. doi.org/10.1186/s13059-026-04167-y | Pubmed
Relevant code:
Spatial Transcriptomics As Rasterized Image Tensors (STARIT) characterizes cell states with subcellular molecular heterogeneity
Dee Velazquez, Caleb Hallinan, Roujin An, Kalen Clifton, Jean Fan^
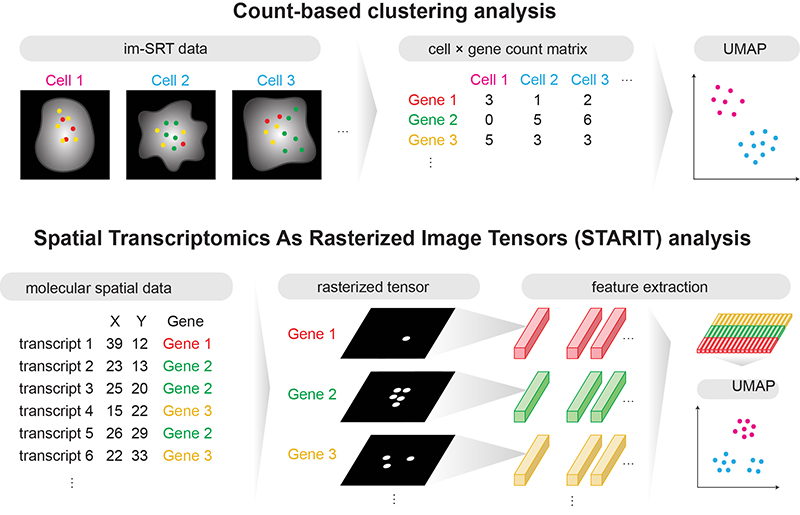
Paper: bioRxiv
Relevant code:
STcompare - comparative spatial transcriptomics data analysis of structurally matched tissues to characterize differentially spatially patterned genes
Kalen Clifton, Vivien Jiang, Rafael dos Santos Peixoto, Srujan Singh, Ryo Matsuura, Hamid Rabb, Jean Fan^
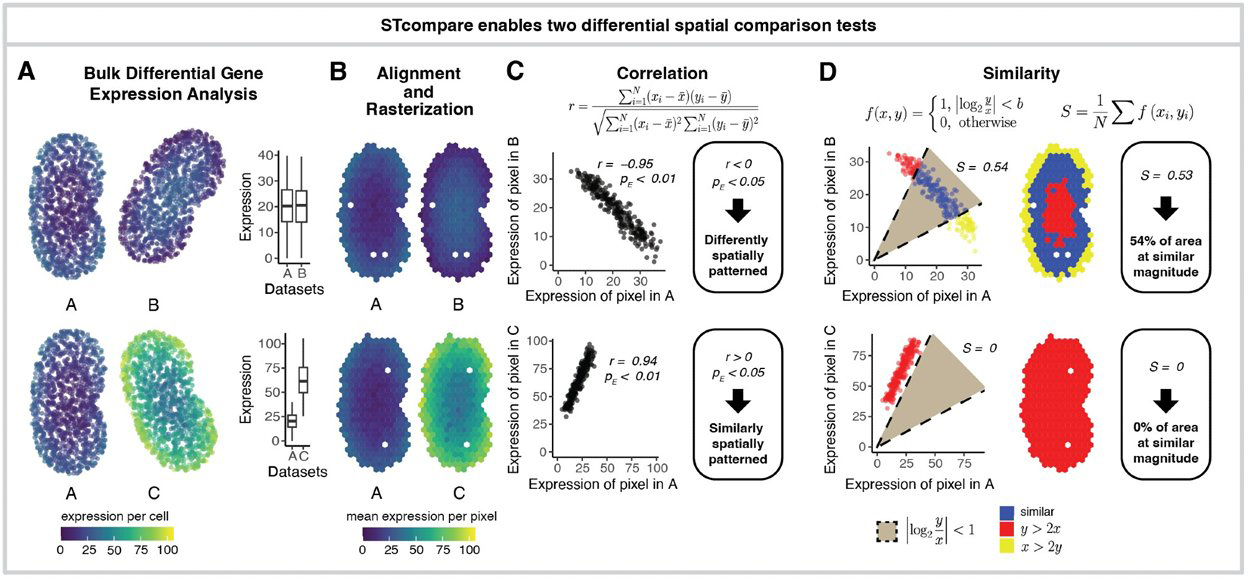
Paper: bioRxiv
Relevant code:
Impact of Data Quality on Deep Learning Prediction of Spatial Transcriptomics from Histology Images
Caleb Hallinan, Calixto-Hope G. Lucas, Jean Fan^

Paper: bioRxiv
Relevant code:
Evidence of off-target probe binding in the 10x Genomics Xenium v1 Human Breast Gene Expression Panel compromises accuracy of spatial transcriptomic profiling
Caleb Hallinan, Hyun Joo Ji, Steven L Salzberg, Jean Fan^
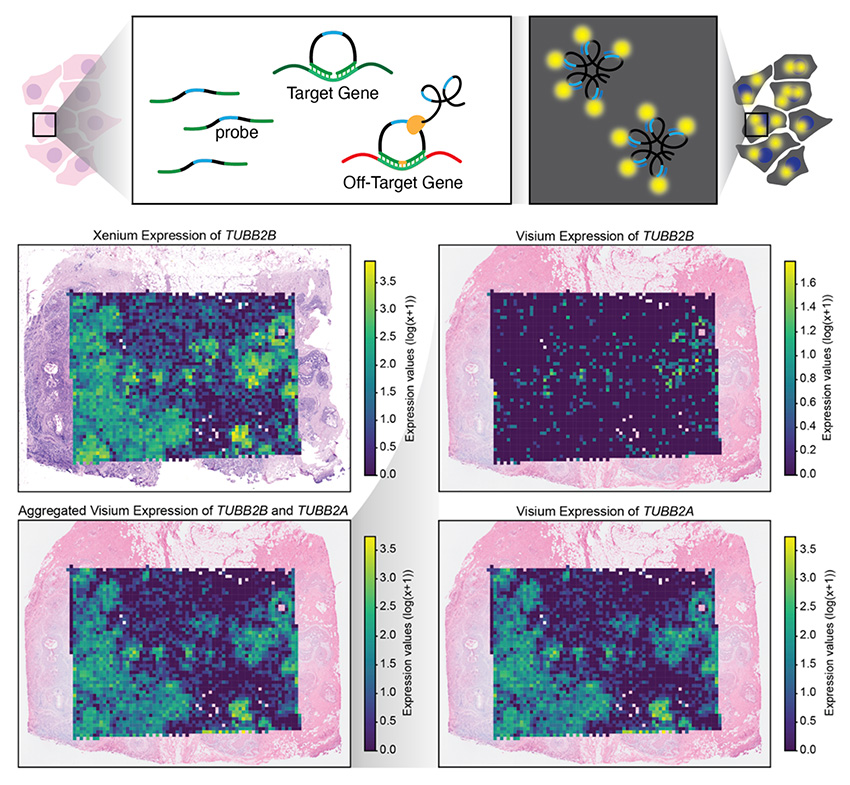
Paper: eLife. August 26, 2025. doi.org/10.7554/eLife.107070.1
Relevant code:
scatterbar - an R package for visualizing proportional data across spatially resolved coordinates
Dee Velazquez, Jean Fan^
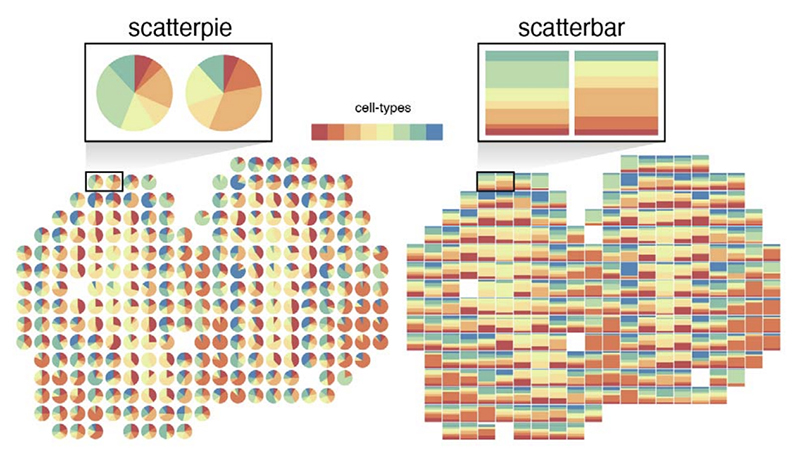
Paper: Bioinformatics, Volume 41, Issue 2, February 2025, btaf047 | Pubmed
Relevant code:
Characterizing cell-type spatial relationships across length scales in spatially resolved omics data
Rafael dos Santos Peixoto, Brendan F. Miller, Maigan A. Brusko, Gohta Aihara, Lyla Atta, Manjari Anant, Mark A. Atikinson, Todd M. Brusko, Clive H. Wasserdall, Jean Fan^

Paper: Nature Communications. January 3, 2025. doi.org/10.1038/s41467-024-55700-1 | Pubmed
Relevant code:
SEraster - a rasterization preprocessing framework for scalable spatial omics data analysis
Gohta Aihara, Kalen Clifton, Mayling Chen, Zhuoyan Li, Lyla Atta, Brendan F. Miller, Rahul Satija, John W Hickey, Jean Fan^
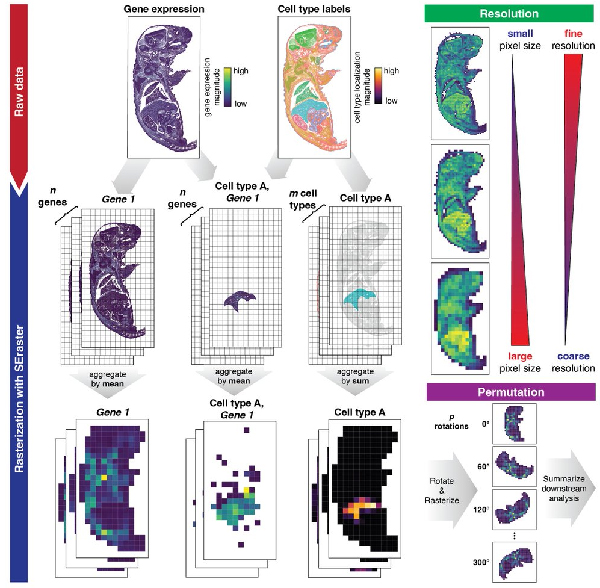
Paper: Bioinformatics. June 20, 2024. doi.org/10.1093/bioinformatics/btae412 | Pubmed
Relevant code:
Gene count normalization in single-cell imaging-based spatially resolved transcriptomics
Lyla Atta, Kalen Clifton, Manjari Anant, Gohta Aihara, and Jean Fan^
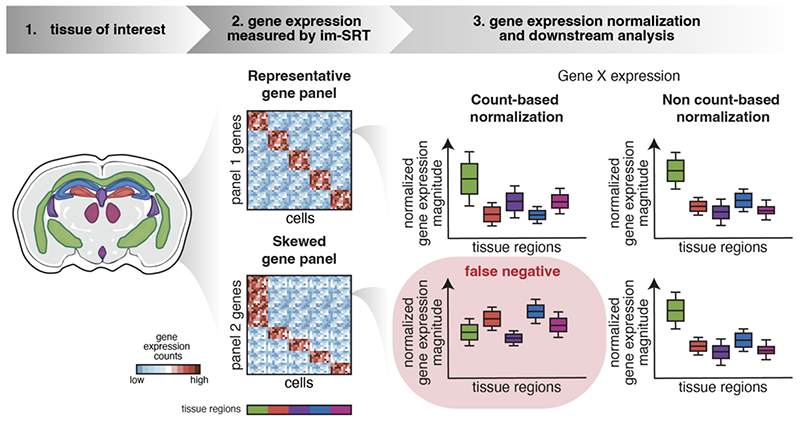
Paper: Genome Biology. June 12, 2024. doi.org/10.1186/s13059-024-03303-w | Pubmed
Relevant code:
Single-cell morphology encodes functional subtypes of senescence in aging human dermal fibroblasts
Pratik Kamat, Nico Macaluso, Chanhong Min, Yukang Li, Anshika Agrawal, Aaron Winston, Lauren Pan, Bartholomew Starich, Teasia Stewart, Pei-Hsun Wu, Jean Fan, Jeremy Walston, Jude M Phillip
Paper: Science Advances | Pubmed
STalign - alignment of spatial transcriptomics data using diffeomorphic metric mapping
Kalen Clifton*, Manjari Anant*, Gohta Aihara, Lyla Atta, Osagie K Aimiuwu, Justus M Kebschull, Michael I Miller, Daniel Tward^, Jean Fan^
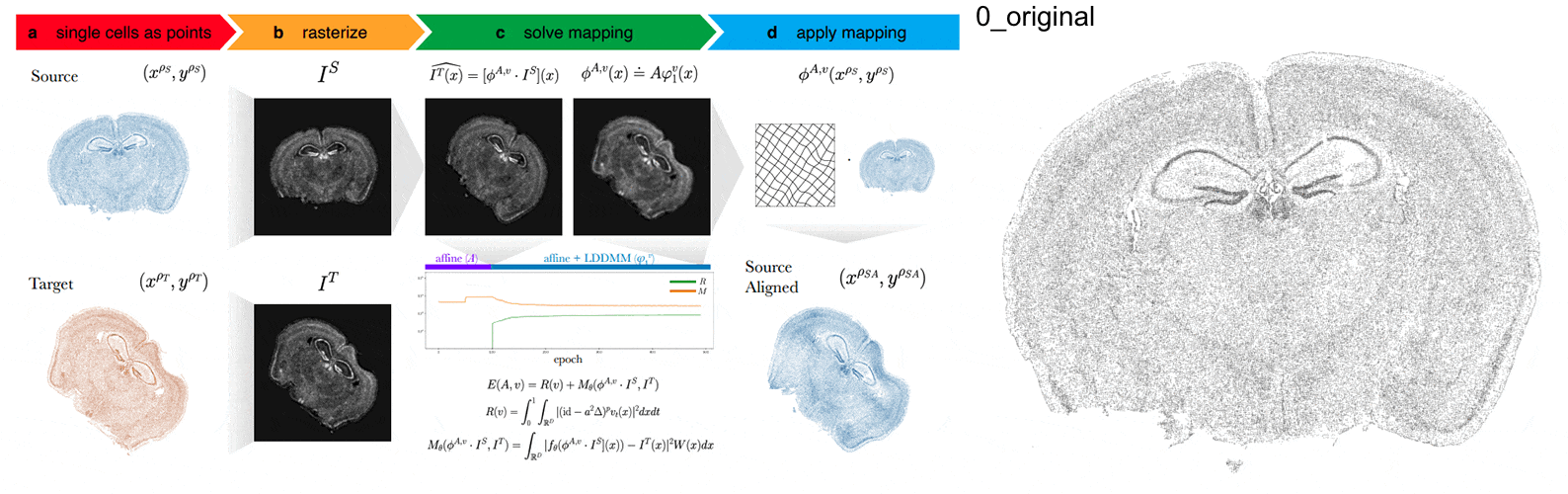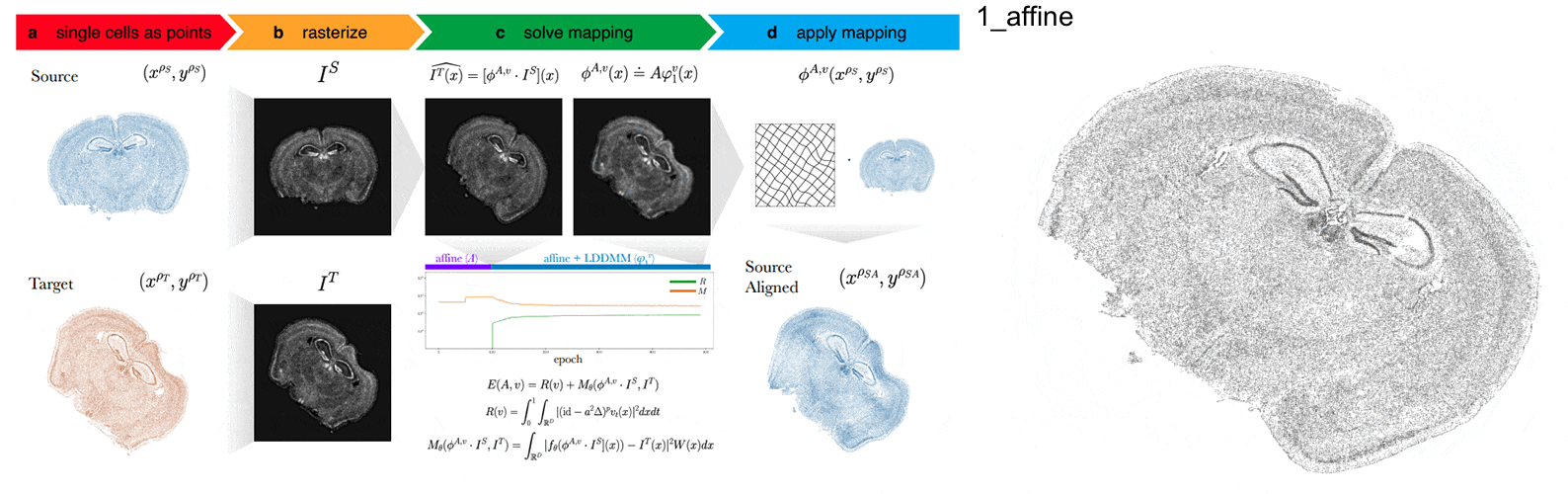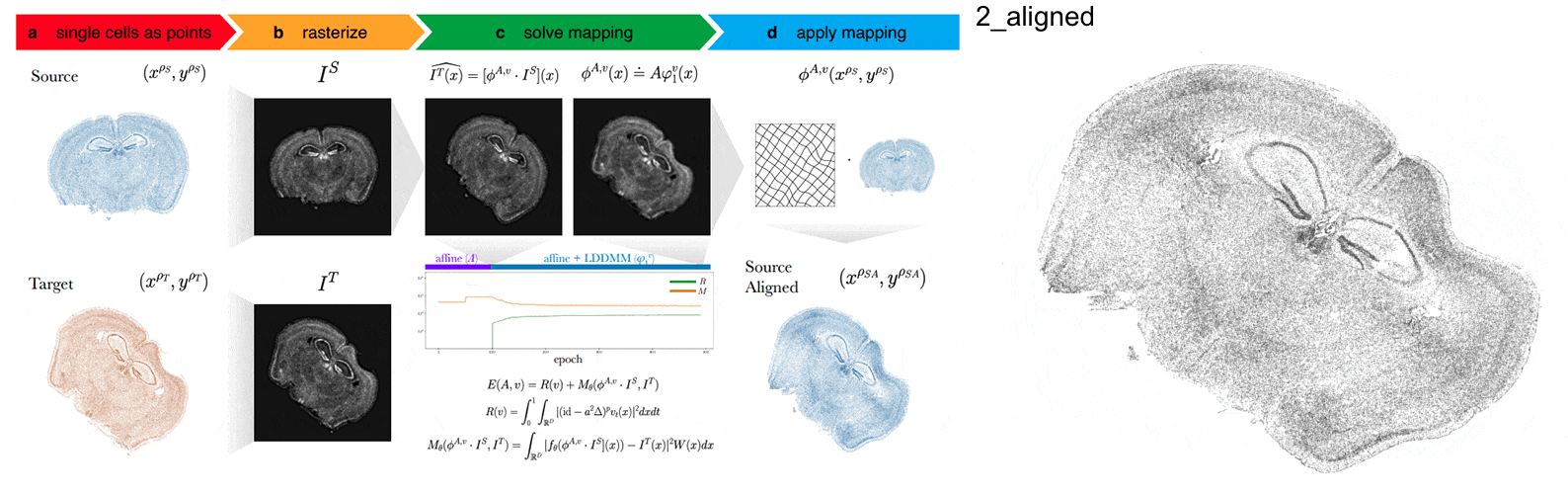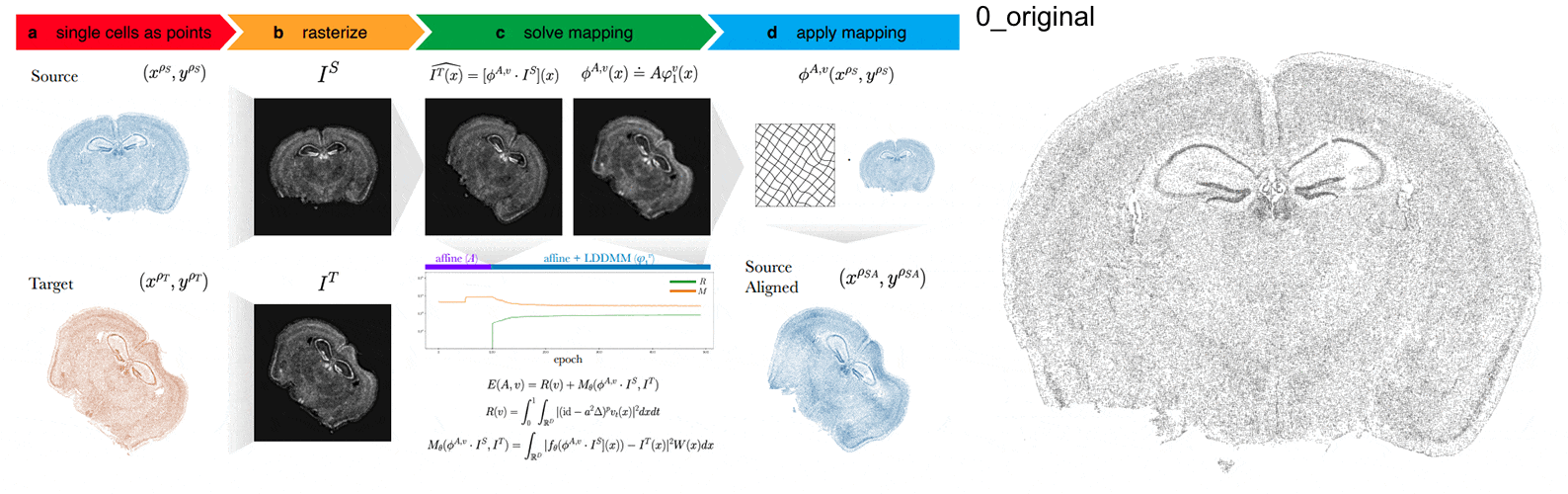
Paper: Nature Communications. December 8, 2023. doi.org/10.1038/s41467-023-43915-7 | Pubmed
Relevant code:
Soluble PD-L1 reprograms blood monocytes to prevent cerebral edema and facilitate recovery after ischemic stroke
Jennifer E Kim, Ryan P Lee, Eli Yazigi, Lyla Atta, James Feghali, Ayush Pant, Aanchal Jain, Idan Levitan, Eileen Kim, Kisha Patel, Nivedha Kannapadi, Pavan Shah, Adnan Bibic, Zhipeng Hou, Justin M Caplan, L Fernando Gonzalez, Judy Huang, Risheng Xu, Jean Fan, Betty Tyler, Henry Brem, Vassiliki A Boussiotis, Lauren Jantzie, Shenandoah Robinson, Raymond C Koehler, Michael Lim, Rafael J Tamargo, Christopher M Jackson
Paper: Brain, Behavior, and Immunity. December 7, 2023. doi.org/10.1016/j.bbi.2023.12.007 | Pubmed
Single cell and spatial transcriptomics analysis of kidney double negative T lymphocytes in normal and ischemic mouse kidneys
Sepideh Gharaie, Kyungho Lee, Kathleen Noller, Emily K. Lo, Brendan Miller, Hyun Jun Jung, Andrea M. Newman-Rivera, Johanna T. Kurzhagen, Nirmish Singla, Paul A. Welling, Jean Fan, Patrick Cahan, Sanjeev Noel, Hamid Rabb
Paper: Scientific Reports. Nov 23, 2023. doi.org/10.1038/s41598-023-48213-2 | Pubmed
Why it's worth making computational methods easy to use
Jean Fan
Paper: Nature. April 27, 2023. doi.org/10.1038/d41586-023-01440-z | Pubmed
Cross-modality mapping using image varifolds to align tissue-scale atlases to molecular-scale measures with application to 2D brain sections
Kaitlin M. Stouffer, Alain Trouvé, Laurent Younes, Michael Kunst, Lydia Ng, Hongkui Zeng, Manjari Anant, Jean Fan, Yongsoo Kim, Xiaoyin Chen, Mara Rue, Michael I. Miller
Paper: Nature Communications. April 25, 2024. doi.org/10.1038/s41467-024-47883-4 | Pubmed
Reference-free cell type deconvolution of pixel-resolution spatially resolved transcriptomics data
Brendan F Miller, Feiyang Huang, Lyla Atta, Arpan Sahoo, Jean Fan^
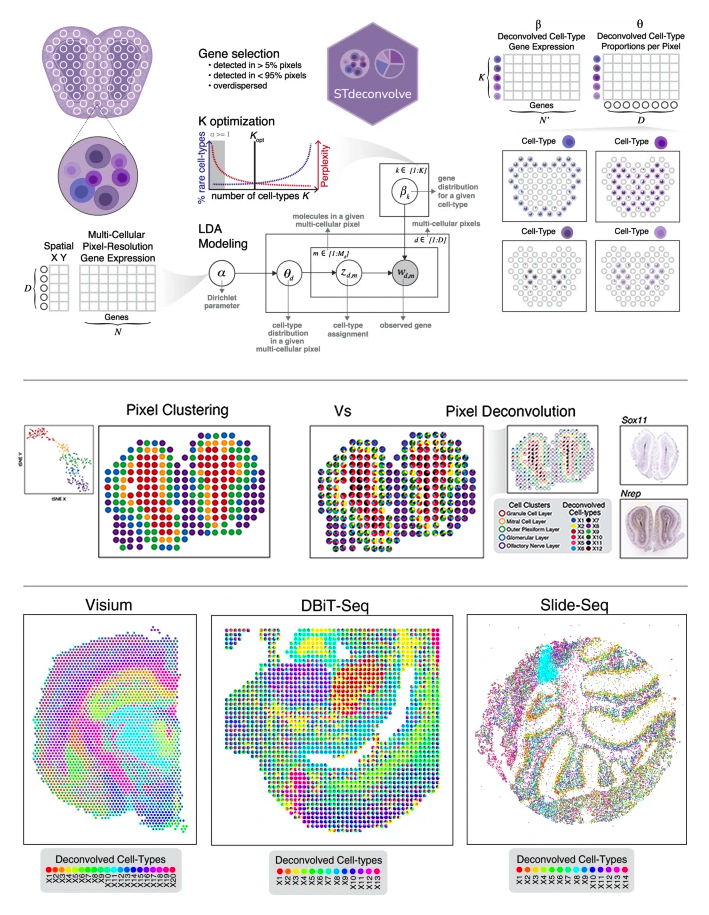
Paper: Nature Communications. April 29, 2022. doi.org/10.1038/s41467-022-30033-z | Pubmed | PDF
Relevant code:
Single cell analysis reveals immune dysfunction from the earliest stages of CLL that can be reversed by ibrutinib
Noelia Purroy Zuriguel, Yuzhou Evelyn Tong, Camilla K Lemvigh, Nicoletta Cieri, Shuqiang Li, Erin M Parry, Wandi Zhang, Laura Z Rassenti, Thomas J Kipps, Susan L Slager, Neil E Kay, Connie Lesnick, Tait D Shanafelt, Paolo Ghia, Lydia Scarfo, Kenneth J Livak, Peter V Kharchenko, Donna Neuberg, Lars Ronn Olsen, Jean Fan, Satyen H Gohil, Catherine J Wu^
Paper: Blood. January 12, 2022. doi.org/10.1182/blood.2021013926 | Pubmed | PDF
VeloViz - RNA-velocity informed embeddings for visualizing cellular trajectories
Lyla Atta, Arpan Sahoo, Jean Fan^
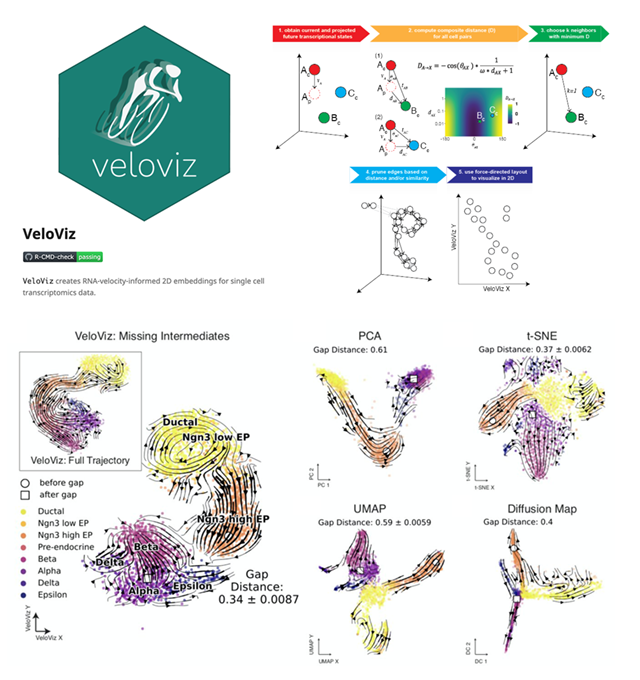
Paper: Bioinformatics. September 28, 2021. doi.org/10.1093/bioinformatics/btab653 | Pubmed | PDF
Relevant code:
Computational challenges and opportunities in spatially resolved transcriptomic data analysis
Lyla Atta, and Jean Fan^
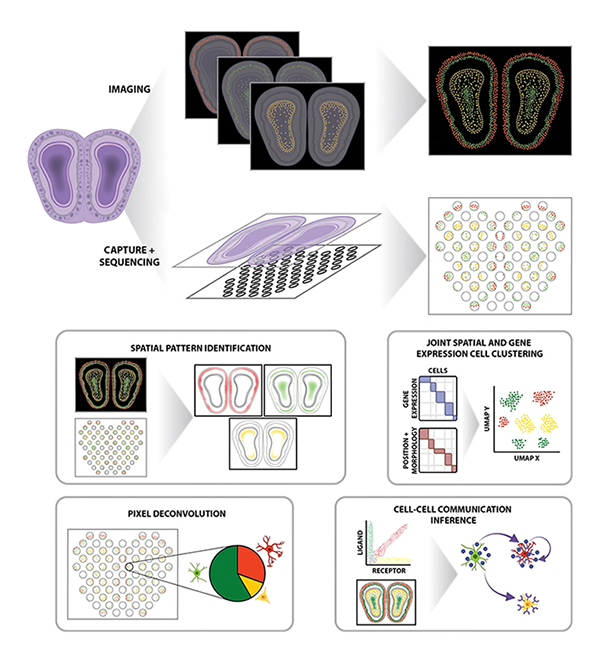
Paper: Nature Communications. September 6, 2021. doi.org/10.1038/s41467-021-25557-9 | Pubmed | PDF
Rewiring of human neurodevelopmental gene regulatory programs by human accelerated regions
Kelly M. Girskis*, Andrew B. Stergachis*, Ellen M. DeGennaro*, Ryan N. Doan, Xuyu Qian, Matthew B. Johnson, Peter P. Wang, Gabrielle M. Sejourne, M. Aurel Nagy, Elizabeth A. Pollina, André M. M. Sousa, Taehwan Shin, Connor J. Kenny, Julia L. Scotellaro, Brian M. Debo, Dilenny M. Gonzalez, Lariza M. Rento, Rebecca C. Yeh, Janet H. T. Song, Marc Beaudin, Jean Fan, Peter V. Kharchenko, Nenad Sestan, Michael E. Greenberg, Christopher A. Walsh^
Paper: Neuron. September 2, 2021. doi.org/10.1016/j.neuron.2021.08.005 | Pubmed | PDF
Multi Scale Diffeomorphic Metric Mapping of Spatial Transcriptomics Datasets
Michael I. Miller, Jean Fan, Daniel J. Tward
Paper: Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR) Workshops
Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma
Toshiro Hara*, Rony Chanoch-Myers*, Nathan D. Mathewson, Chad Myskiw, Lyla Atta, Lillian Bussema, Stephen W. Eichhorn, Alissa C. Greenwald, Gabriela S. Kinker, Christopher Rodman, L. Nicolas Gonzalez Castro, Hiroaki Wakimoto, Orit Rozenblatt-Rosen, XiaoweiZhuang, Jean Fan, Tony Hunter, Inder M. Verma, Kai W. Wucherpfennig, Aviv Regev, Mario L. Suvà^, Itay Tirosh^
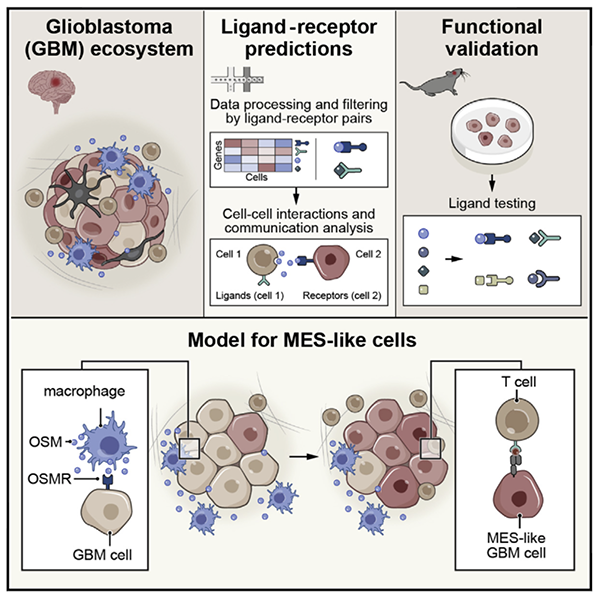
Paper: Cancer Cell. June 3, 2021. doi:10.1016/j.ccell.2021.05.002 | Pubmed | PDF
Characterizing spatial gene expression heterogeneity in spatially resolved single-cell transcriptomics data with nonuniform cellular densities
Brendan F Miller, Dhananjay Bambah-Mukku, Catherine Dulac, Xiaowei Zhuang, Jean Fan^
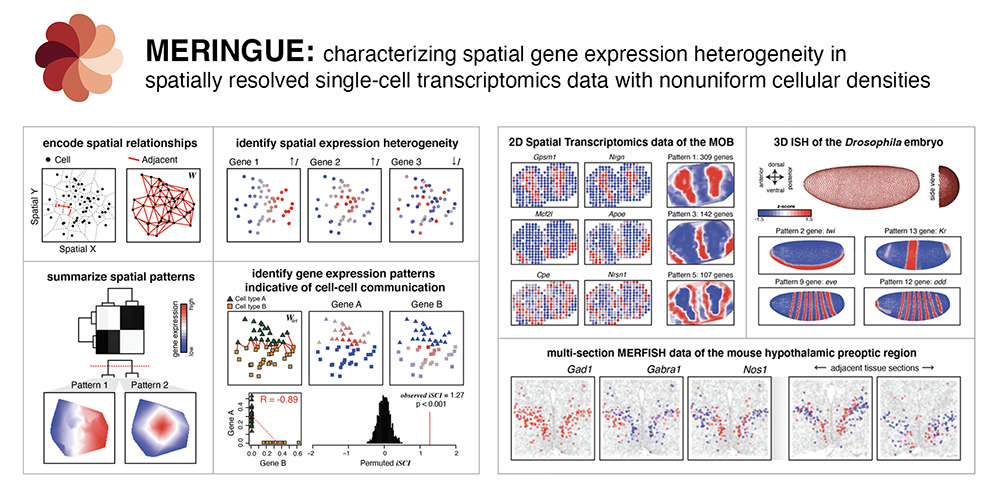
Paper: Genome Research. Advance May 25, 2021, doi:10.1101/gr.271288.120 | Pubmed | PDF
Relevant code:
Spatial organization of the transcriptome in individual neurons
Guiping Wang, Cheen-Euong Ang, Jean Fan, Andrew Wang, Jeffrey R. Moffitt, Xiaowei Zhuang^
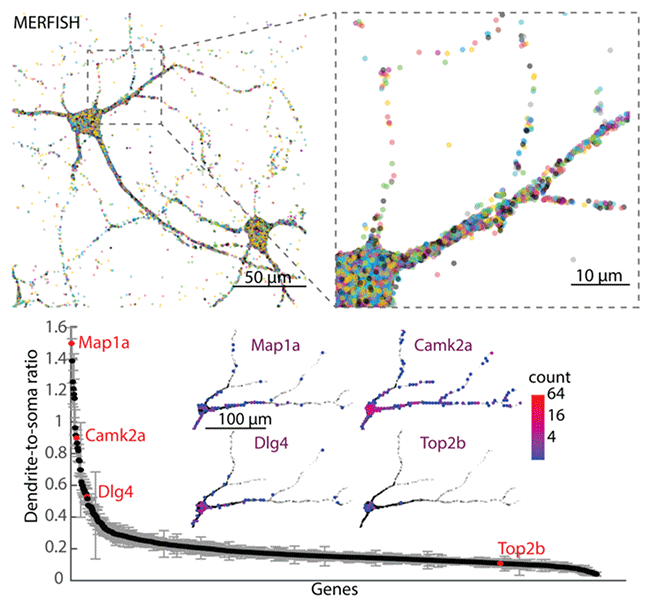
Paper: bioRxiv
Single-cell transcriptomics in cancer - computational challenges and opportunities
Jean Fan^, Kamil Slowikowski, Fan Zhang
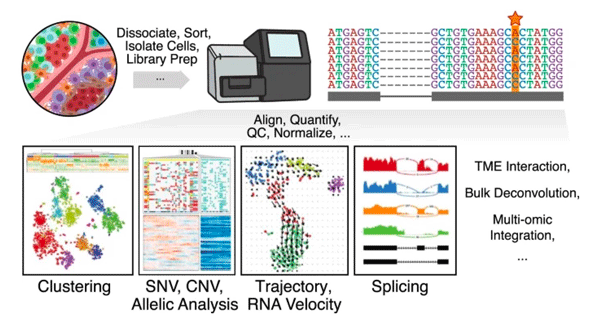
Paper: Nature Experimental and Molecular Medicine. Sept 15, 2020, doi.org:10.1038/s12276-020-0422-0 | Pubmed | PDF
Fast, sensitive and accurate integration of single-cell data with Harmony
Ilya Korsunsky, Nghia Millard, Jean Fan, Kamil Slowikowski, Fan Zhang, Kevin Wei, Yuriy Baglaenko, Michael Brenner, Po-ru Loh, and Soumya Raychaudhuri^
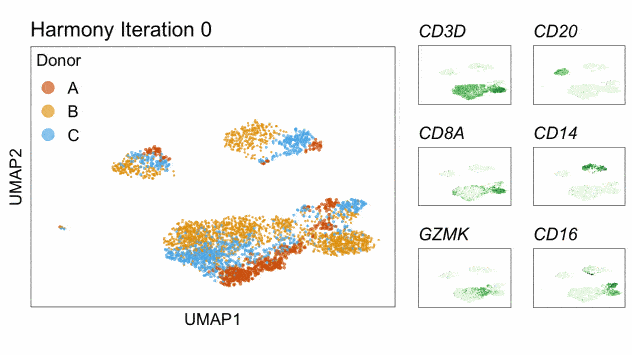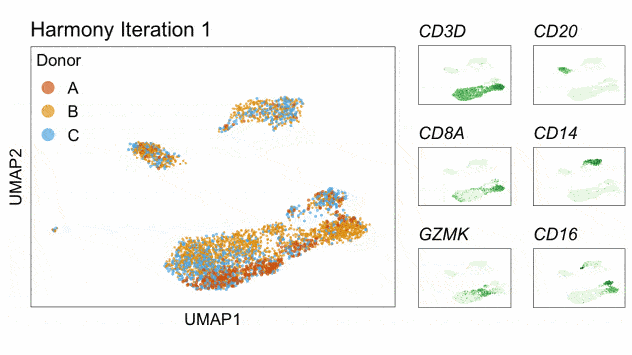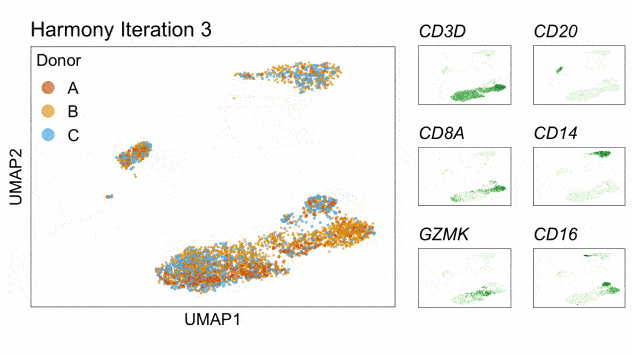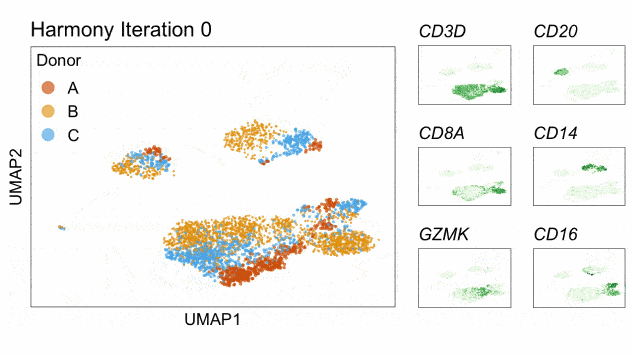
Paper: Nature Methods. November 18, 2019. doi:10.1038/s41592-019-0619-0 | Pubmed | PDF
Relevant code:
Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression
Chenglong Xia*, Jean Fan*, George Emanuel*, Junjie Hao, and Xiaowei Zhuang^
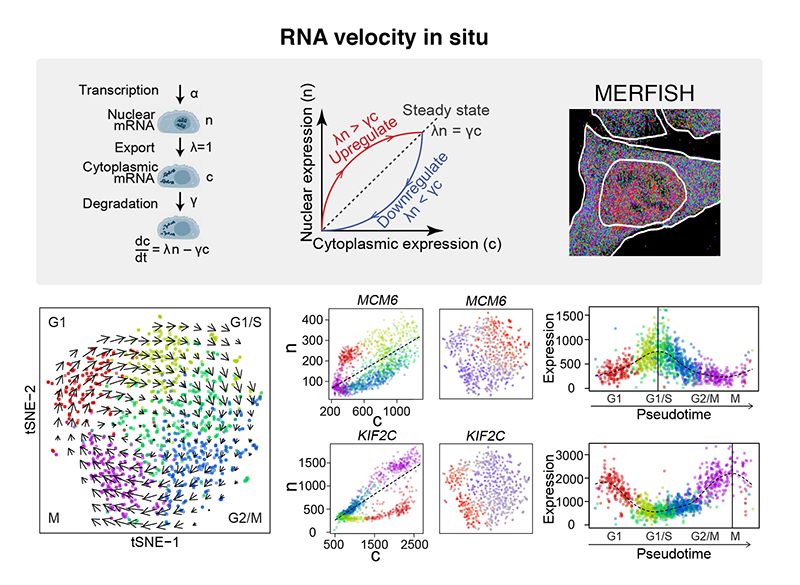
Paper: PNAS. Sept 9, 2019, doi:10.1073/pnas.1912459116 | Pubmed | PDF
A Murine Model of Chronic Lymphocytic Leukemia Based on B Cell-Restricted Expression of Sf3b1 Mutation and Atm Deletion
Shanye Yin, Rutendo G. Gambe, Jing Sun, Aina Zurita Martinez, Zachary J.Cartun, Fara Faye D.Regis, Youzhong Wan, Jean Fan, Angela N.Brooks, Sarah E. M. Herman, Elisaten Hacken, AmaroTaylor-Weiner, Laura Z. Rassenti, Emanuela M. Ghia, Thomas J. Kipps, Esther A. Obeng, Carrie L. Cibulskis, Donna Neuberg, Dean R.Campagna, Mark D. Fleming, Benjamin L. Ebert, Adrian Wiestner, Ignaty Leshchiner, James A. DeCaprio, Gad Getz, Robin Reed, Ruben D. Carrasco, Catherine J. Wu^, Lili Wang^
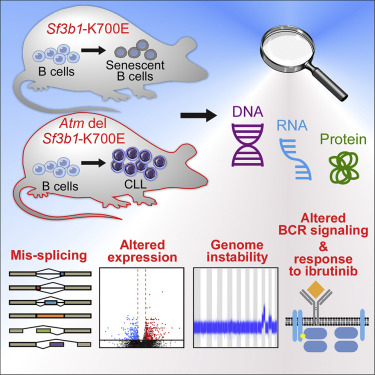
Paper: Cancer Cell. Feb 11, 2019. doi:10.1016/j.ccell.2018.12.013 | Pubmed | PDF
RNA velocity of single cells
Gioele La Manno, Ruslan Soldatov, Amit Zeisel, Emelie Braun, Hannah Hochgerner, Viktor Petukhov, Katja Lidschreiber, Maria E Kastriti, Peter Lönnerberg, Alessandro Furlan, Jean Fan, Lars E Borm, Zehua Liu, David van Bruggen, Jimin Guo, Xiaoling He, Roger Barker, Erik Sundström, Gonçalo Castelo-Branco, Patrick Cramer, Igor Adameyko, Sten Linnarsson^, Peter V Kharchenko^

Paper: Nature. August 8, 2018. doi:10.1038/s41586-018-0414-6 | Pubmed | PDF
Relevant code:
Linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell RNA-seq data
Jean Fan*, Hae-Ock Lee*, Soohyun Lee, Da-eun Ryu, Semin Lee, Catherine Xue, Seok Jin Kim, Kihyun Kim, Nikolas Barkas, Peter J Park, Woong-Yang Park^ and Peter V Kharchenko^
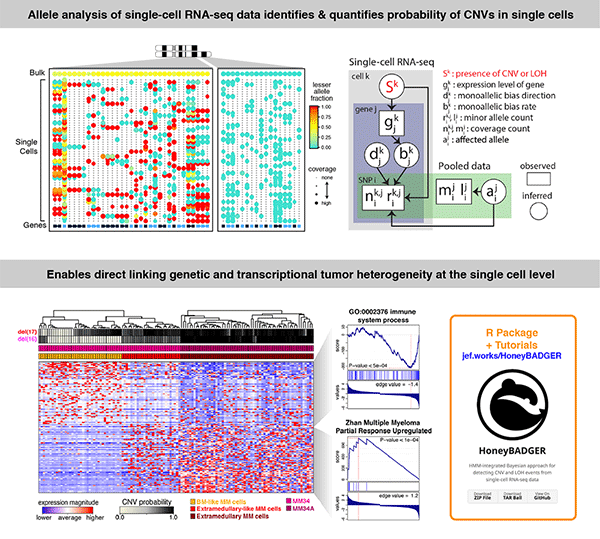
Paper: Genome Research. June 13, 2018. doi:10.1101/gr.228080.117 | Pubmed | PDF
Relevant code:
Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain
Blue B Lake*, Song Chen*, Brandon C Sos*, Jean Fan*, Gwendolyn E Kaeser, Yun C Yung, Thu E Duong, Derek Gao, Jerold Chun^, Peter V Kharchenko^, Kun Zhang^
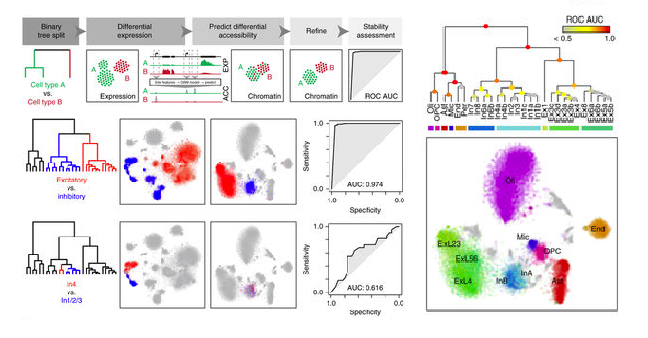
Paper: Nature Biotechnology. December 11, 2017. doi:10.1038/nbt.4038 | Pubmed | PDF
Relevant code:
Integrated single-cell genetic and transcriptional analysis suggests novel drivers of chronic lymphocytic leukemia
Lili Wang*, Jean Fan*, Joshua M. Francis, George Georghiou, Sarah Hergert, Shuqiang Li, Rutendo Gambe, Chensheng W. Zhou, Chunxiao Yang, Sheng Xiao, Paola Dal Cin, Michaela Bowden, Dylan Kotliar, Sachet A. Shukla, Jennifer R. Brown, Donna Neuberg, Dario R. Alessi, Cheng-Zhong Zhang^, Peter V. Kharchenko, Kenneth J. Livak, Catherine J. Wu^
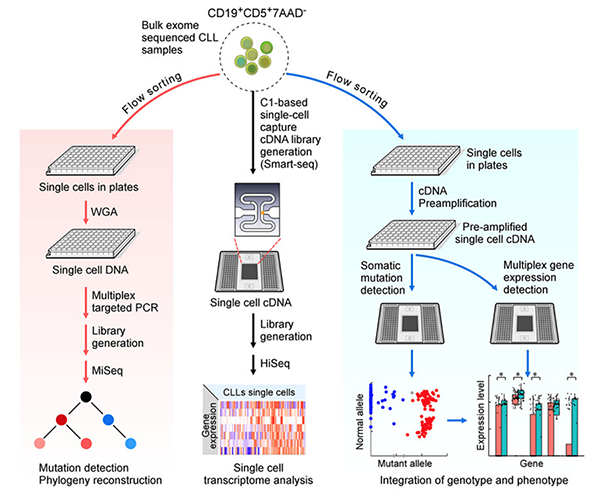
Paper: Genome Research. May 22, 2017. doi/10.1101/gr.217331.116 | Pubmed | PDF
Transcriptomic Characterization of SF3B1 Mutation Reveals Its Pleiotropic Effects in Chronic Lymphocytic Leukemia
Lili Wang*, Angela N. Brooks*, Jean Fan*, Youzhong Wan*, Rutendo Gambe, Shuqiang Li, Sarah Hergert, Shanye Yin, Samuel S. Freeman, Joshua Z. Levin, Lin Fan, Michael Seiler, Silvia Buonamici, Peter G. Smith, Kevin F. Chau, Carrie L. Cibulskis, Wandi Zhang, Laura Z. Rassenti, Emanuela M. Ghia, Thomas J. Kipps, Stacey Fernandes, Donald B. Bloch, Dylan Kotliar, Dan A. Landau, Sachet A. Shukla, Jon C. Aster, Robin Reed, David S. DeLuca, Jennifer R. Brown, Donna Neuberg, Gad Getz, Kenneth J. Livak, Matthew M. Meyerson, Peter V. Kharchenko, Catherine J. Wu^
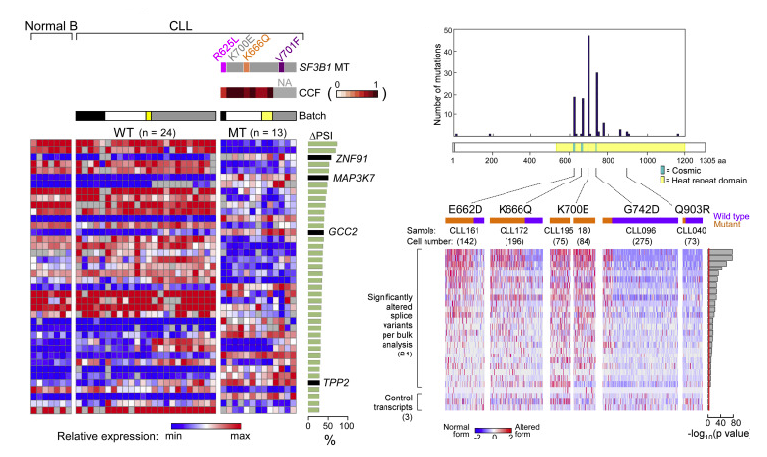
Paper: Cancer Cell. November 3, 2016. doi.org/10.1016/j.ccell.2016.10.005 | Pubmed | PDF
Cell-Type-Specific Alternative Splicing Governs Cell Fate in the Developing Cerebral Cortex
Xiaochang Zhang^, Ming Hui Chen, Xuebing Wu, Andrew Kodani, Jean Fan, Ryan Doan, Manabu Ozawa, Jacqueline Ma, Nobuaki Yoshida, Jeremy F. Reiter, Douglas L. Black, Peter V. Kharchenko, Phillip A. Sharp, Christopher A. Walsh^
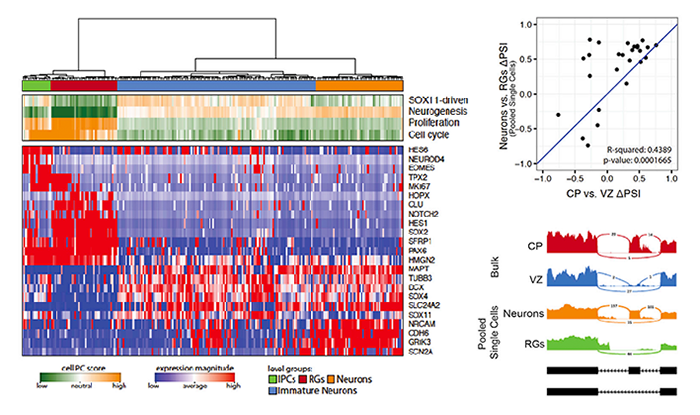
Paper: Cell. August 25, 2016. doi.org/10.1016/j.cell.2016.07.025 | Pubmed | PDF
Relevant code:
Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition
Jan A. Burger*^, Dan A. Landau*, Amaro Taylor-Weiner*, Ivana Bozic*, Huidan Zhang*, Kristopher Sarosiek, Lili Wang, Chip Stewart, Jean Fan, Julia Hoellenriegel, Mariela Sivina, Adrian M. Dubuc, Cameron Fraser, Yulong Han, Shuqiang Li, Kenneth J. Livak, Lihua Zou, Youzhong Wan, Sergej Konoplev, Carrie Sougnez, Jennifer R. Brown, Lynne V. Abruzzo, Scott L. Carter, Michael J. Keating, Matthew S. Davids, William G. Wierda, Kristian Cibulskis, Thorsten Zenz, Lillian Werner, Paola Dal Cin, Peter Kharchencko, Donna Neuberg, Hagop Kantarjian, Eric Lander, Stacey Gabriel, Susan O’Brien, Anthony Letai, David A. Weitz, Martin A. Nowak, Gad Getz, Catherine J. Wu^
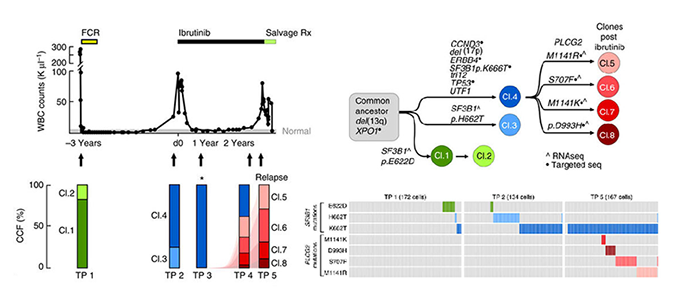
Paper: Nature Communications. May 20, 2016. doi:10.1038/ncomms11589 | Pubmed | PDF
Relevant code:
Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis
Jean Fan, Neeraj Salathia, Rui Liu, Gwendolyn E Kaeser, Yun C Yung, Joseph L Herman, Fiona Kaper, Jian-Bing Fan, Kun Zhang, Jerold Chun, Peter V Kharchenko^
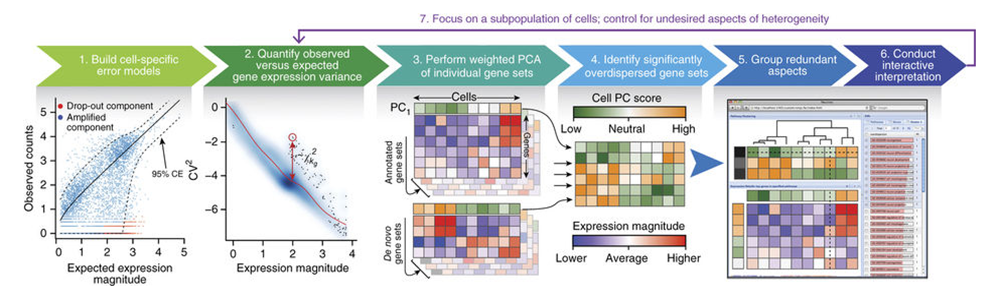
Paper: Nature Methods. Jan 18, 2016. doi:10.1038/nmeth.3734 | Pubmed | PDF
Relevant code:
Locally Disordered Methylation Forms the Basis of Intratumor Methylome Variation in Chronic Lymphocytic Leukemia
Dan A. Landau*, Kendell Clement*, Michael J. Ziller, Patrick Boyle, Jean Fan, Hongcang Gu, Kristen Stevenson, Carrie Sougnez, Lili Wang, Shuqiang Li, Dylan Kotliar, Wandi Zhang, Mahmoud Ghandi, Levi Garraway, Stacey M. Fernandes, Kenneth J. Livak, Stacey Gabriel, Andreas Gnirke, Eric S. Lander, Jennifer R. Brown, Donna Neuberg, Peter V. Kharchenko, Nir Hacohen, Gad Getz, Alexander Meissner, Catherine J. Wu^
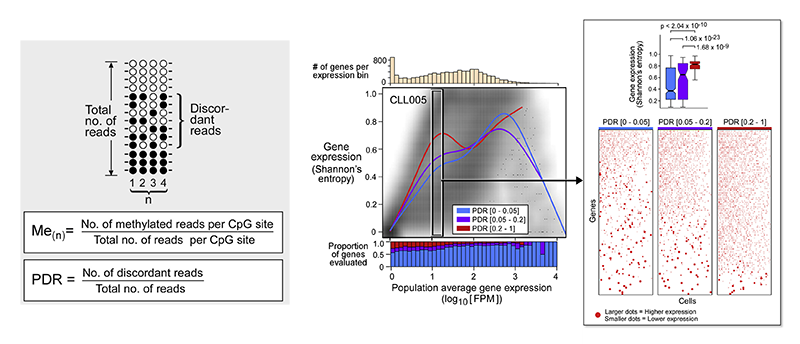
Paper: Cancer Cell. Dec 8, 2014. doi:10.1016/j.ccell.2014.10.012 | Pubmed | PDF